

Abstract
OBJECTIVE To study the microsatellite instability (MSI) and methylation state of the hMLHI gene promoter and their mechanisms underlying the development of gastric cancer.
METHODS Forty-one gastric cancer samples were obtained from patients undergoing surgery and 46 chronic atrophic gastritis tissues with dysplasia or intestinal metaplasia (IM) were obtained from patients undergoing gastro-endoscopy. Fourteen normal gastric mucosal samples were used as controls. Genomic DNA was extracted from the samples and 5 microsatellite markers were used to measure MSI. Methylation - specific PCR (MSP) was used to screen the methylation state of the samples. DNA sequencing and immunohistochemistry were performed to verify the results.
RESULTS MSI was identified in 22 out of the 41 (53.7%) gastric cancers, of which 8 cases showed high-level MSI (2 or more loci altered) and 14 showed low-level MSI (1 locus altered). MSI was also detected in 12 out of 46 (26.1%) pre-cancerous lesions of the stomach, whereas it was not seen in the normal tissue. Moreover, hMLHI hypermethylation was detected in 17 out of the 41 (41.5%) gastric cancers, 9 out of the 46 (19.6%) pre-cancerous lesions and 0 out of the 14 normal tissue. Significant differences in frequency of MSI and hMLHI promoter methylation were observed among gastric cancers, precancerous lesions and normal gastric tissue. Gastric samples with MSI had a tendency to be hypermethylated in the hMLHI promoter. DNA sequencing and immunohistochemistry results also confirmed that hMLHI promoter methylation could lead to loss of the hMLHI protein and gene silence which sequentely resulted in gene mismatch and MSI.
CONCLUSION Accumulation of MSI and hMLHI promoter methylation may be important early molecular events during gastric carcinogenesis and may contribute to the acquisition of a transformed cell phenotype and the development of gastric cancer.
keywords
Epigenetic changes have been reported in many cancers and are now recognized to be at least as common as genetic changes.[1] Aberrant methylation of cytosine located within the dinucleotide CpG is by far the best-categorized epigenetic change. The cancer cell genome shows global hypomethylation as well as regional promoter hypermethylation of several tumor suppressor genes.[2] Promoter hypermethylation is an alternative mechanism of gene inactivation in carcinogenesis. Several studies have suggested that hypermethylation of the promoter is associated with loss of gene expression and is observed in neoplasia. By inactivating various tumor suppressor genes, this epigenetic modification can affect many important cellular processes, such as the cell cycle (pl4, pl5, pl6 genes), DNA repair (hMLH1, hMSH2, BRCA1) and the metastasizing process (E-cadherin, TIMP3).[3]
Gastric cancer is the most common cancer worldwide and the leading cancer in several countries. Its etiology is unknown, but gastric mucosal infection by the bacterium Helicobacter pylori and environmental factors have been implicated in gastric carcinogenesis. Because of difficulties in early diagnosis and poor efficacy of treatment, the 5-year survival rate of gastric cancer is <10%.[4] Human gastric cancer is believed to develop through a multi-step process which involves basal cell hyperplasia, chronic atrophic gastritis, intestinal metaplasia, dysplasia, carcinoma in situ and advanced carcinoma. Understanding of the molecular mechanisms in this process will not only provide biomarkers for early diagnosis, but help us to improve treatment modalities. Our current understanding of gastric carcinogenesis is based on the development of genomic instability. One form of genomic instability, MSI, has been identified in gastric tumor. Among human sporadic tumors, gastric carcinoma possesses the highest prevalence of MSI, with up to 30% of cases manifesting this abnormality.[5]
MSI comprises length mutations in tandem oligonucleotide repeats that occur in a large subset of human tumors.[5] This type of mutation is believed to be a failure of the DNA-MMR system which cannot correct errors that occur during the replication of DNA. This leads to the accumulation of nucleotide mutations and alterations in the length of microsatellite sequences that occur throughout the genome. MSI represents a hypermutable phenotype and correlates with the absence of either hMLH1 or hMSH2.[5] The hMLH1 protein, a mismatch repair enzyme, maintains the fidelity of the genome during cellular proliferation. It acts as a ‘molecular matchmaker’, recruiting other DNA-repair proteins to the mismatch repair complex.[5] Dysfunction of a mismatch repair system could alter microsatellites. [5] In sporadic endometrial carcinomas, loss of hMLH1 expression is frequently the result of hypermethylation of hMLH1, whereas hypermethylation of the hMSH2 promoter region in cancer was not found. Furthermore, reversal of methylation by treatment of cells with 5-aza-2’-deoxycytidine can restore the expression of the hMLH1 protein and MMR capability. Taken together, these findings suggest a possible mechanism by which failure of MMR occurs in these tumors.
To clarify the possible involvement of hMLH1 in gastric carcinogenesis, in this study we sought to evaluate the rate of MSI, the expression of the hMLH1, and the methylation status of the hMLH1 promoter in gastric cancers, pre-cancerous lesions and normal tissue of the stomach.
Materials and Methods
Stomach specimens and DNA extraction
Forty-one gastric cancer samples were obtained from patients undergoing surgery and 46 chronic gastritis samples with dysplasia or intestinal metaplasia were acquired from patients undergoing gastro-endoscopy in the General Hospital, Tianjin, China. Fourteen normal gastric mucosal samples used as controls, were obtained from deceased people undergoing autopsy. The sections from each specimen were examined by 2 pathologists. The tumors were characterized according to Lauren’s histologic clhssification into intestinal or diffuse-type gastric cancer. Lesions of chronic gastritis, intestinal metaplasia and dysplasia were graded as mild, moderate and severe according to the WHO criteria. The clinical characteristics of the patients studied are shown in Table 1. The fresh samples were collected at the time of surgery and frozen at -80°C. Genomic DNA was isolated by standard proteinase-K digestion and a phenol-chloroform extraction protocol as previously described.[1]
Methylation-specific PCR for the hMLH1 promoter
The promoter methylation status of the hMLH1 gene was determined by methylation-specific PCR (MSP), which was described previously.[5] MSP distinguishes unmethylated from hypermethylated alleles in a given gene based on sequence changes produced after bisulfite treatment of DNA, which converts unmethylated, but not methylated, cytosines to uracils. Briefly, 1 μg of DNA was denatured with NaOH and treated with sodium bisulfite. DNA samples were then purified using a Wizard DNA Purification Resin (Promega), treated again with NaOH, precipitated with ethanol, and resuspended in TE buffer. The dissolved DNA was amplified by PCR using primers specific for the methylated (M) or unmethylated (UM) sequences. Two to 3 μl of template were amplified in a thermocycler (PE9700) as follows: 5 min of denaturation at 95°C, then 35 cycles consisting of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s. PCR products were separated on 2% agar gels containing ethidium bromide.
DNA sequencing
Before the MS-PCR products were sequenced by cycle sequencing, a PCR purification kit (Promega) was used to remove unwanted reagents from the PCR reaction. The purified PCR products were then directly cycle-sequenced using a Beckman CEQ 2000 according to the manufacturer’s instructions.
Determination of microsatellite instability (MSI)
Microsatellite analysis was performed on the tumors, precancerous lesions and corresponding normal DNA using a panel of 5 National Cancer Institute workshop-recommended markers.[6] The primer sequences for 5 microsatellite loci are shown in Table 2.[6] Two microliters of PCR product was electrophoresed on 8% denaturing polyacrylamide gels containing 6 mol/L urea at room temperature followed by silver staining. Samples producing PCR products of abnormal sizes in electrophoretic mobility at 2 or more of the 5 loci analyzed were considered as MSI-high (MSI-H), whereas those showing a shift in one locus were classified as MSI-low (MSI-L). The remaining samples, lacking MSI events, were determined to be microsatellite stable (MSS).
Immunohistochemistry
Immunostaining for hMLH1 was performed on all of the samples using the standard streptavidin-biotin-per-oxidase complex method as described previously. [7] Tissue sections (6 μm) which had been fixed in 10% neutral-buffered formalin and paraffin embedded were incubated for 1 h at 37°C using a monoclonal antibody. Microwave pretreatment at 95°C for 30 min in citrate buffer (pH 6.0) was performed after deparaffinization. The stains were graded: (a) negative when there was complete absence of staining in the tissue cell nuclei; and (b) positive when there was positive staining in the tissue cell nuclei.
Statistical analysis
Statistical analysis was performed using the χ2 or Fisher’s Exact test. P<0.05 was considered statistically significant.
Results
Msi
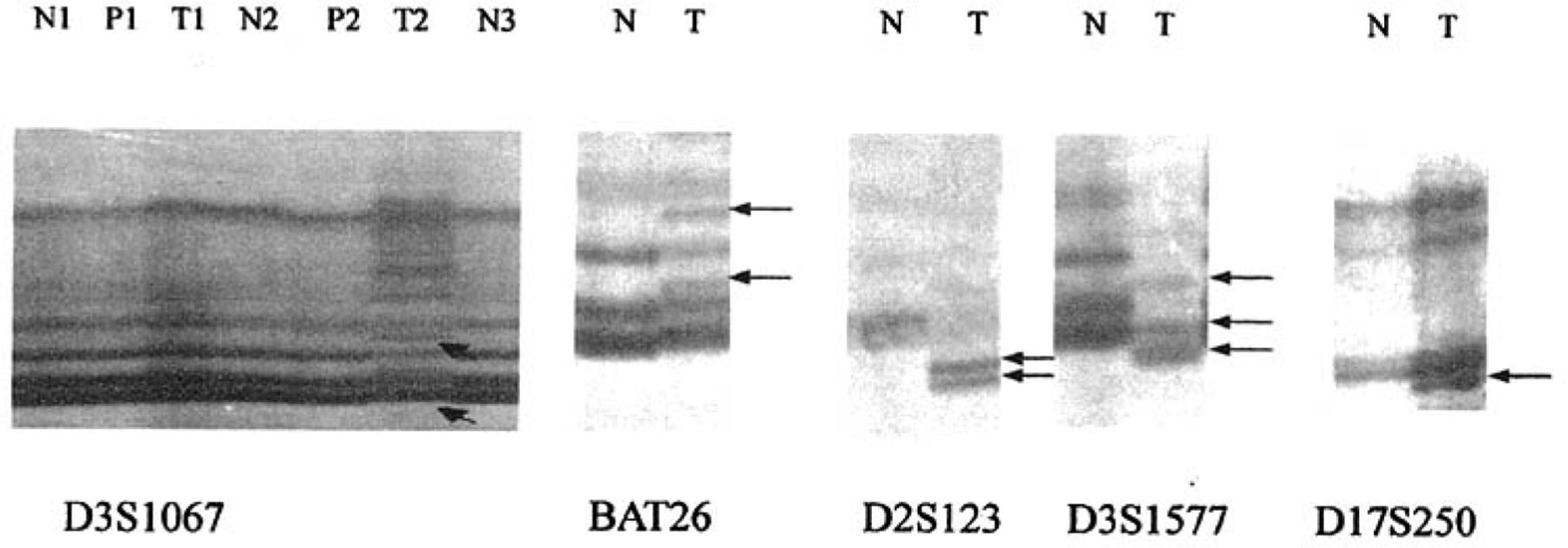
Of the 41 patients with gastric cancer, 22 (53.7%) were MSI, 8(19.5%) were MSI-H and 14(34.1%) were MSI-L(Table 3). However only 12 (26.1%) were MSI in patients with gastric precancerous lesions, of which 6 (13%) were MSI-H. None were MSI in the control group. Significant differences in frequency of MSI were observed among gastric cancers, precancerous lesions and normal mucosal samples (P<0.05, Table 3, Fig.1).
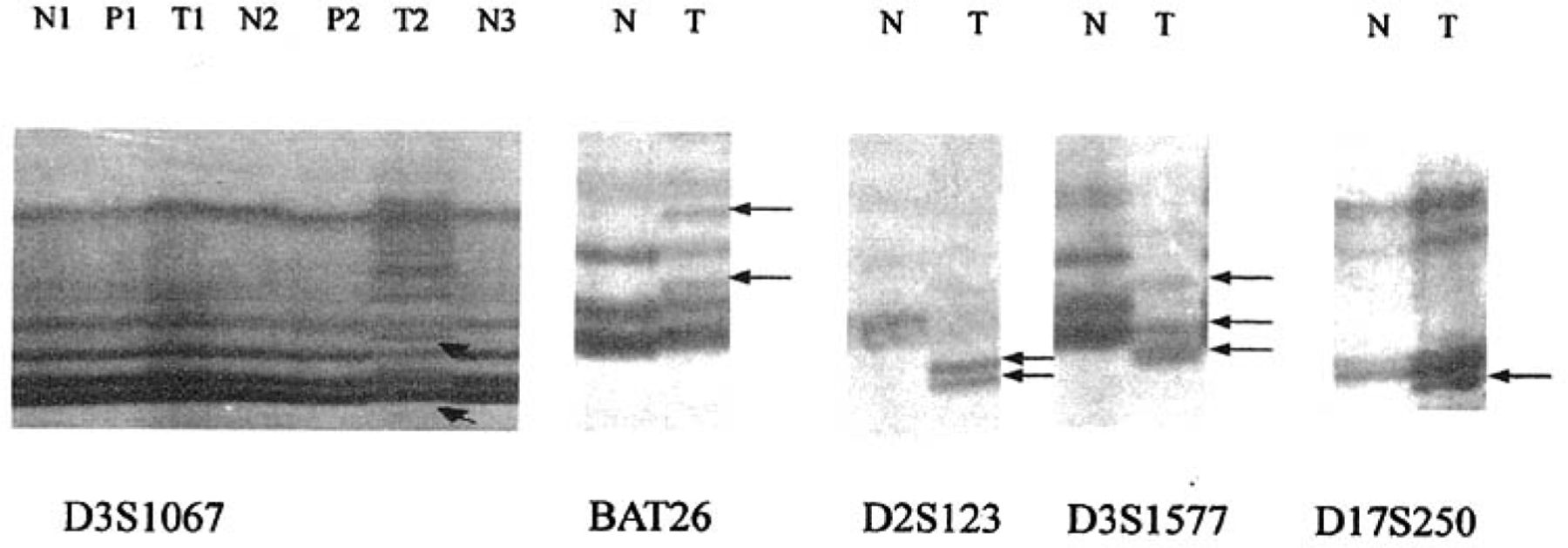
Detection of microsatellite instability (MSI) with five markers in normal gastric tissue, gastric cancer and precancerous lesions. Arrows show abnormal bond of MSI. T: tumor; P: Precancerous lesions; N: normal mucosa.
Immunohistochemistry
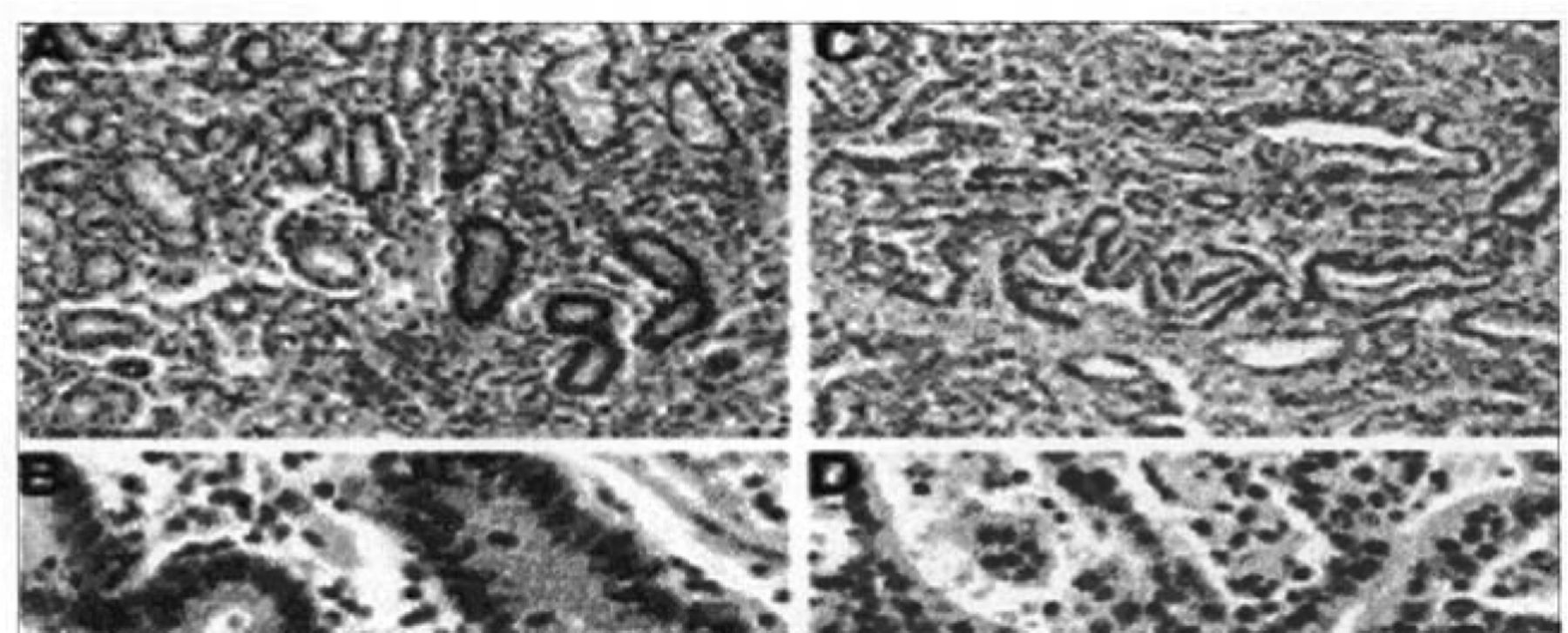
HMLHl staining of the non-neoplastic tissues was consistent with previous reports showing strong positive nuclear staining. In gastric samples, decreased hMLH1 expression was noted in 10 of 14 MSI-H cases with hypermethylation and 9 of 20 MSI-L cases, but none of the MSS cases showed lower hMLH1 protein expression (Table 4, Fig.2). It is notable that the hMLH1 protein was still expressed in the metaplasia, dysplasia, and carcinoma in situ but was completely absent in the invasive part of the same tumor case. Microsatellite analysis of DNA extracted from the dysplastic and carcinoma in situ components in this case showed a microsatellite stable phenotype. Thus, the onset of MSI correlated with the loss of the hMLH1 protein.
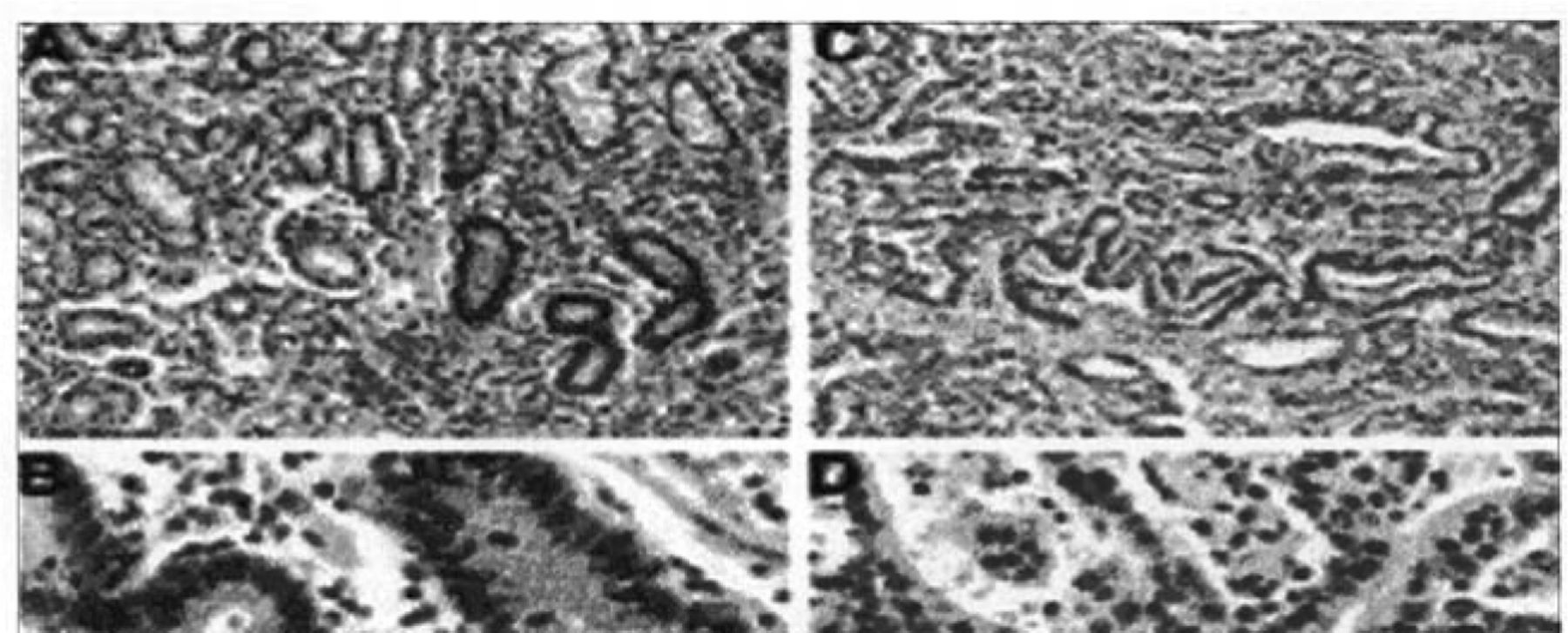
Immunohistochemical results of hMLH1 levels in gastric samples. Views (A and B) of normal gastric mucosa expressing abundant hMLH1 protein in nuclei are shown. Views (C and D) of gastric cancer with MSI and hypermethylation of hMLH1, showing absence of hMLH1 protein in neoplastic nuclei but its abundance in surrounding normal nuclei.
hMLH1 promoter methylation analysis
Results of hMLH1 promoter hypermethylation assays versus MSI studies in samples are summarized in Tables 3, 4 and Figs.3, 4. Using MSP, we demonstrated that hMLH1 gene hypermethylation occurred in 17 of 41(41.5%) gastric cancers, 9 of 46 (19.6%) gastric pre-cancerous lesions and 0 (0%) of 14 normal control samples. All of the 14 MSI-H cases were consistent with hMLH1 promoter methylation. Ten of these 14 cases had loss of the hMLH1 protein. Eleven of 20 (55%) MSI-L demonstrated hMLH1 hypermethylation. Taking these 2 subgroups together, 25 of 34 cases (73.5%) showing either low or high MSI were hypermethylated. In contrast, only 1 of 67 MSI-negative patients (1.5%) exhibited hMLH1 promoter hypermethylation (P<0.05 for MSI-H plus MSI-L versus MSI-negative, Fisher’s Exact test, 2 tailed). The association of MSI-H with hMLH1 promoter methylation and hMLH1 protein loss was statistically highly significant using the Fisher’s Exact test (P<0.05). Hence, the samples with MSI generally showed higher methylation frequencies than did the MSS samples. In most cases, methylation of the target genes was biallelic, but in some cases, an unmethylated signal was visible, presumably as a result of monoallelic methylation or normal mesenchymal cells, such as lymphocytes or fiber cells.
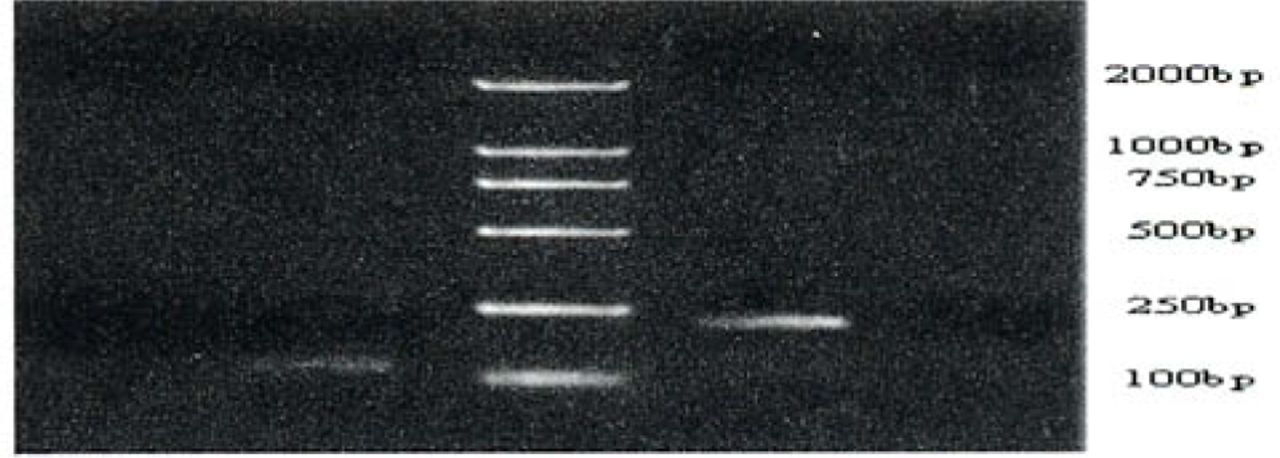
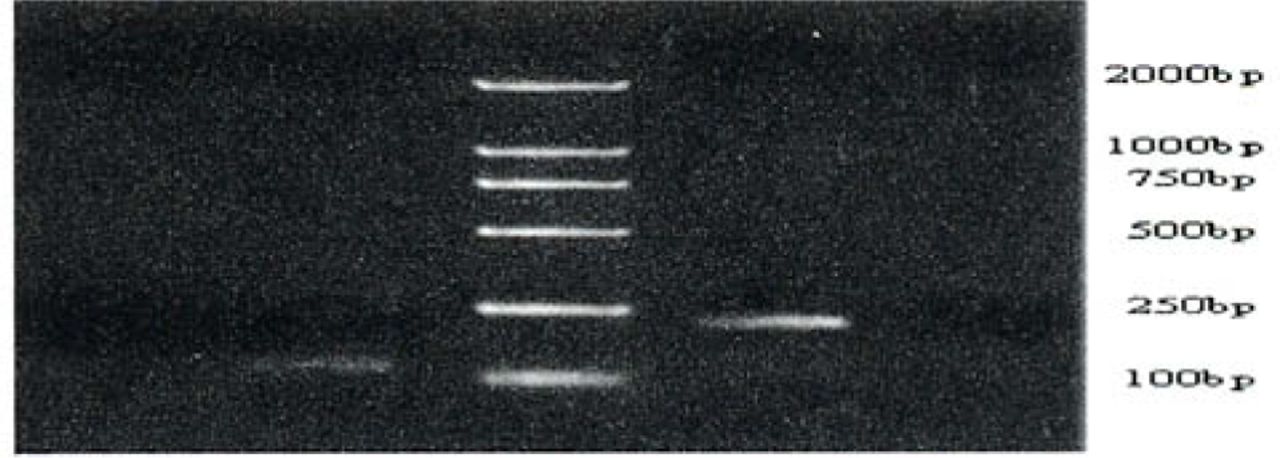
Methylation-specific PCR of the hMLH1 promoter region in gastric cancers, precancerous lesions and normal gastric tissue. Bisulfite-treated DNA was amplified with methylated and unmethylated specific hMLH1 primers. The 124-bp product is indicative of an unmethylated hMLH1 allele, whereas the 196-bp product indicates a methylated hMLH1 allele. T and N respectively shows products amplified from the gastric cancers and normal gastric mucosa tissue. MP and UMP stand for products amplified using specific methylated and unmethylated primers.

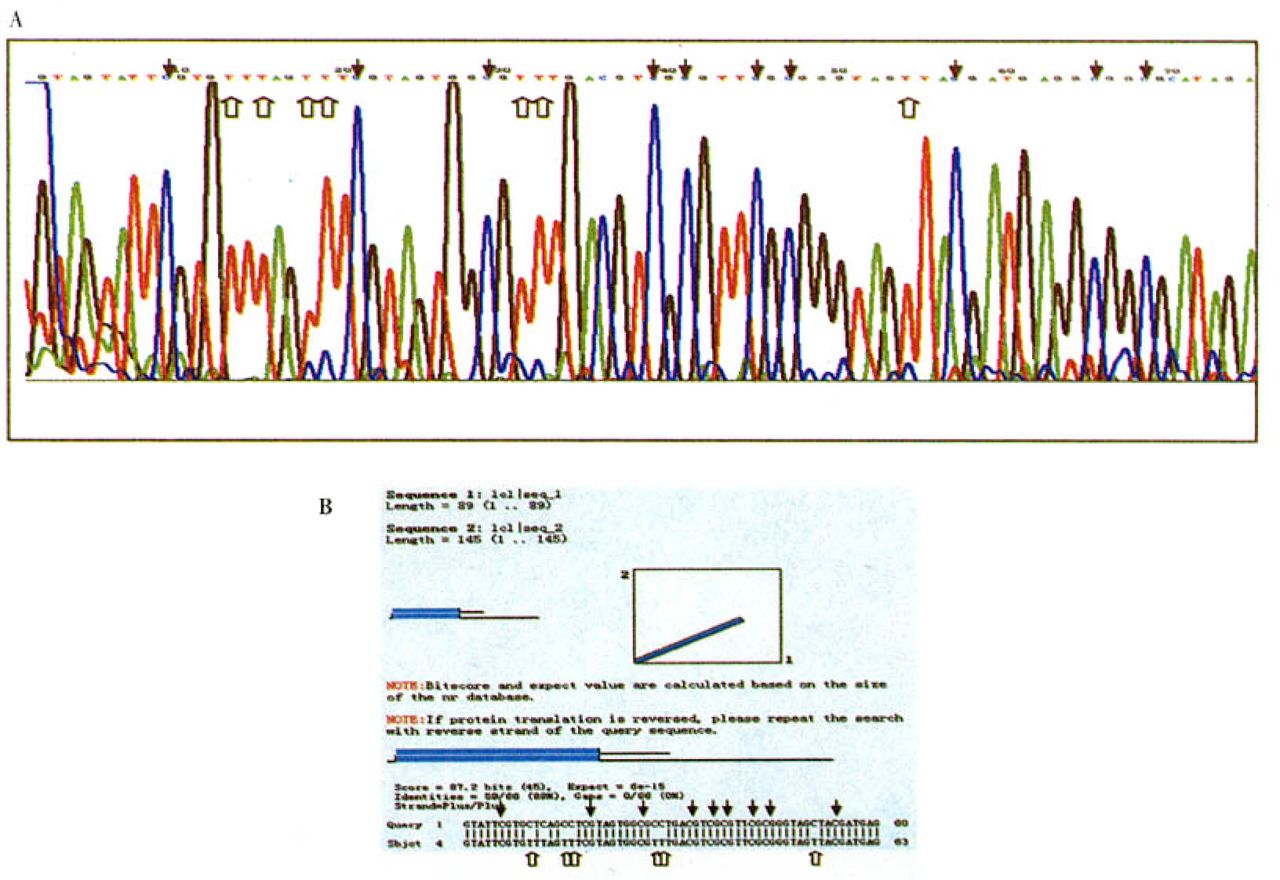
A: Sequencing histograms (matching GenBank accession No. U26559) of entirely methylated hMLH1 promoter region amplified with sodium bisulfite-treated genomic DNAs from a primary gastric carcinoma. B: The sequencing result of methylated hMLH1 compares with the original hMLH1 sequence in NCBI Blast bank. Black arrows show that methylated CpG sites still appear as CpG, while white arrows show that unmethylated simple cytosines appear as thymines in the final sequence.
Direct sequencing of the methylated DNA PCR product confirmed the retention of cytosines at all CpGs within the PCR product, whereas single cytosines were all converted to thymines in the DNA-PCR product (Fig.4).
Discussion
Genetic and epigenetic changes in tumor associated genes (TAGs) are inevitable events in the initiation and progression of human cancer.[1] Since gene promoter hypermethylation and chromatin structure alteration are comparable with genetic mutations or deletions of tumor suppressor genes (TSGs) in cancer, it is very urgent to find some target genes that are inactivated by hypermethylation. In fact there are many examples of CpG methylation-mediated transcriptional silencing of TSGs, such as pl6, pl5, APC, Rb, hMLH1, E-cad etc.[3] In our study we have found that hMLH1 is a target gene for hypermethylation in gastric carcinogenesis. This aberrant methylation can cause cancer by inactivation of the hMLH1 gene and lead to a series of molecular events.
Our data document the importance of aberrant methylation of the hMLH1 promoter in causing MSI in sporadic gastric cancer. MSI occurs in more than 50% of sporadic gastric carcinomas and is significantly more common in samples displaying hMLH1 promoter hypermethylation. hMLH1 methylation occurs in gastric carcinoma at a high frequency (41.5%). In contrast, MSI and hMLH1 methylation were detected at a much lower frequency in a group of precancerous lesions and even cannot be detected in a single case in normal gastric tissue samples (P<0.05). Especially, the data show a significant relationship between hMLH1 hypermethylation and MSI-H gastric carcinogenesis. MSI-H is exclusively associated with hMLH1 hypermethylation: i.e., there are no cases of MSI-H occurring in the absence of HMLHl hypermethylation. These results suggest that hMLH1 hypermethylation and MSI are very important mechanisms in gastric carcinogenesis, which could be good molecular markers to help distinguish cancers from non-neoplastic tissue. At present the difficulty of early histopathological diagnosis in gastric cancers, especially in borderline tumors or intermediate lesions, has been pointed out so that it is urgent to find some molecular biological cancer markers for early diagnosis. Detection of MSI and hMLH1 hypermethylation may be useful as potential diagnostic markers because nearly half of the gastric carcinomas shows these features. Moreover, exploration of the role of epigenetic silencing in gastric carcinogenesis may permit targeted analysis of putative precursor lesions in order to predict malignant potential and prognosis.
Perhaps the most striking finding in our research is that considerable hMLH1 hypermethylation and MSI were found in all 6 precancerous lesion samples taken 5 cm away from the gastric carcinomas, whereas the frequency of hMLH1 hypermethylation and MSI in simple gastric precancerous lesions taken from non-neoplastic patients was much lower. Frequent hMLH1 hypermethylation in non-neoplastic mucosa adjacent to gastric cancers strongly suggests that hypermethylation of hMLH1 is an initial vital event in early tumorigenesis. The phenomenon shows that the lesions with hMLH1 hypermethylation and MSI may be more prone to develop carcinoma. lino et al.[8] have also reported on serrated adenomas with hMLH1 hypermethylation leading to MSI-H cancer. In the colon, considerable hMLH1 promoter hypermethylation in non-neoplastic mucosa adjacent to the tumor also was reported. Given the high prevalence of hMLH1 methylation in gastric precancerous lesions adjacent to the cancers, we can infer that these parts of tissues later will develop the same cancer because they have the same epigenetic and genetic changes. Taken together, these findings suggest that epigenetic silencing of HMLHl may play an important role in progression of precancerous lesions, which, after subsequent inactivation of hMLH1, is required for entry into the pathway leading to microsatellite-unstable gastric cancer. We can conclude that hMLH1 hypermethylation may represent an early event in gastric oncogenesis that culminates in disordered DNA mismatch repair, which precedes other molecular changes and makes it to be a good carcinoma marker for early diagnosis.
To verify our methylation results, we also examined the hMLH1 protein expression level using immunohistochemistry in 3 groups resulting in finding a good correlation. The majority of gastric carcinomas with hMLH1 promoter hypermethylation displayed decreased protein expression and MSI, supporting hypermethylation as the cause of transcriptional inactivation and MMR deficiency. The remaining reduced protein expression in the hMLH1 methylation cases is probably derived from the mesenchyma or admixed normal cells within the tumor. The above data are consistent with previous reports of hMLH1 hypermethylation in colorectal cancers with MSI and support the idea that hMLH1 hypermethylation is the most prevalent mechanism of MMR deficiency.[5] Herman et al.[9] also reported that some colorectal cancer cell lines analyzed showed a direct mechanistic connection between hMLH1 promoter hypermethylation and DNA-MMR deficiency in carcinogenesis, in which methylation of a CpG upstream was associated with complete transcriptional blockade.
Although we cannot completely exclude the possibility of mutations being the cause for the loss of the hMLH1 protein and MSI phenotype in some of these cases with promoter methylation, this possibility is not likely because promoter methylation is widely known to cause MMR-gene transcriptional silencing, while mutations have rarely been described in gastric cancer. Therefore, we believe that hMLH1 hypermethylation is more frequent than mutations in gastric carcinogenesis. Other possible mechanisms underlying MMR deficiency in the MSI-positive gastric tumors:(l) hypermethylation of other known MMR genes; (2) point mutations in known MMR genes; or (3) mutations of as yet unidentified MMR genes.
Our data document that the loss of the hMLH1 protein is a significant event operating in tumor initiation, which also indicates that hMLH1 promoter hypermethylation is associated with hMLH1 transcriptional inactivation and protein reduction. The preservation of the hMLH1 protein in normal gastric tissue, and its absence in some intestinal metaplasia, dysplasia, and carcinoma in situ phase are associated with onset of MSI, which suggests that there is rapid clonal expansion and tumor formation once the hMLH1 protein is lost. MMR deficiency can also accelerate mutations of other important growth-regulatory genes such as transforming growth factor β type II receptor.
Another interesting feature of the present data was the occurrence of methylated and unmethylated PCR products at the same time in some cases with MSS or MSI-L. These samples indicate that early hypermethylation may be partial or monoallelic and MSI-H only happens at the later stage of complete (or biallelic) hypermethylation. The maintenance of MMR proficiency in the partial hypermethylation suggests that hypermethylation of both hMLH1 alleles is required to produce the MSI phenotype. In the MMR-deficient cell lines SW48 and RKO, which have no contaminated normal cells, only methylated hMLH1 PCR product can be detected.[9] In our study none of the MSS gastric samples showed loss of the hMLH1 protein, which supports that the MSS cases had clinicopathological features indicating less potential for cancer formation. Similar observations were noted in a few cases of MSI-L pre-cancerous lesions which are indistinguishable from the MSS cases in clinicopathological features, and they have normal hMLH1 expression. It is possible that a part of these cases are less likely to develop gastric cancer. We also found that several hMLH1 hypermethylated cases had a normal hMLH1 protein level, which supports that partially hypermethylated hMLH1 can still manifest MMR proficiency. Another reason is that even if biallelic methylation exists it is not sufficient for gene silencing because the density of CpG island methylation correlates with the degree of gene inactivation. Fleisher et al.[10] also reported that hypermethylation alone, if not associated with hMLH1 protein reduction, did not cause MSI. These results are consistent with those of previous studies.[5] They demonstrated that only enough (biallelic or complete) hypermethylation of hMLH1 promoter caused protein reduction and sequentially induced MSI.[10] Moreover, de-methylation of the hMLH1 in SW48 and RKO cells can restore MMR proficiency and hMLH1 protein expression, although this treatment achieves only partial demethylation, which again suggests that biallelic hypermethylation is required for MMR inactivation.
Finally, our data showed hypermethylation in 11 of 20 MSI-L cases. We suggest that in MSI-L gastric samples, hypermethylation represents a recent event: hMLH1 hypermethylation initiates MSI, but leaving a temporal window during which some cases have not yet accumulated large numbers of microsatellite instability, which means that the early gastric cancer can be detected by MSI analysis. This theory may also explain the single case in our study in which hMLH1 hypermethylation occurred with MSS and normal hMLH1 expression. The low frequency of such cases suggests that this temporal window is brief.
Taken together we can conclude that hypermethylation frequently targets HMLHl and correlates well with its transcriptional silencing, diminished hMLH1 expression and MSI in gastric cancer, suggesting that in gastric carcinogenesis an epigenetic mechanism underlies hMLH1 gene inactivation and MMR deficiency. Detection of hMLH1 promoter methylation and reduced hMLH1 protein may be useful both as diagnostic markers and for early detection of gastric cancer.
- Received November 25, 2005.
- Accepted March 27, 2006.
- Copyright © 2006 by Tianjin Medical University Cancer Institute & Hospital and Springer
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.