

Abstract
Among the numerous oncogenes involved in human cancers, KRAS represents the most studied and best characterized cancer-related genes. Several therapeutic strategies targeting oncogenic KRAS (KRASonc) signaling pathways have been suggested, including the inhibition of synthetic lethal interactions, direct inhibition of KRASonc itself, blockade of downstream KRASonc effectors, prevention of post-translational KRASonc modifications, inhibition of the induced stem cell-like program, targeting of metabolic peculiarities, stimulation of the immune system, inhibition of inflammation, blockade of upstream signaling pathways, targeted RNA replacement, and oncogene-induced senescence. Despite intensive and continuous efforts, KRASonc remains an elusive target for cancer therapy. To highlight the progress to date, this review covers a collection of studies on therapeutic strategies for KRAS published from 1995 to date. An overview of the path of progress from earlier to more recent insights highlight novel opportunities for clinical development towards KRASonc-signaling targeted therapeutics.
keywords
- Direct inhibition
- downstream effectors
- oncogenic KRAS
- drug target sites
- small GTPases
- signal transduction
- targeting synthetic, lethal interactions
- therapeutic strategies
Introduction
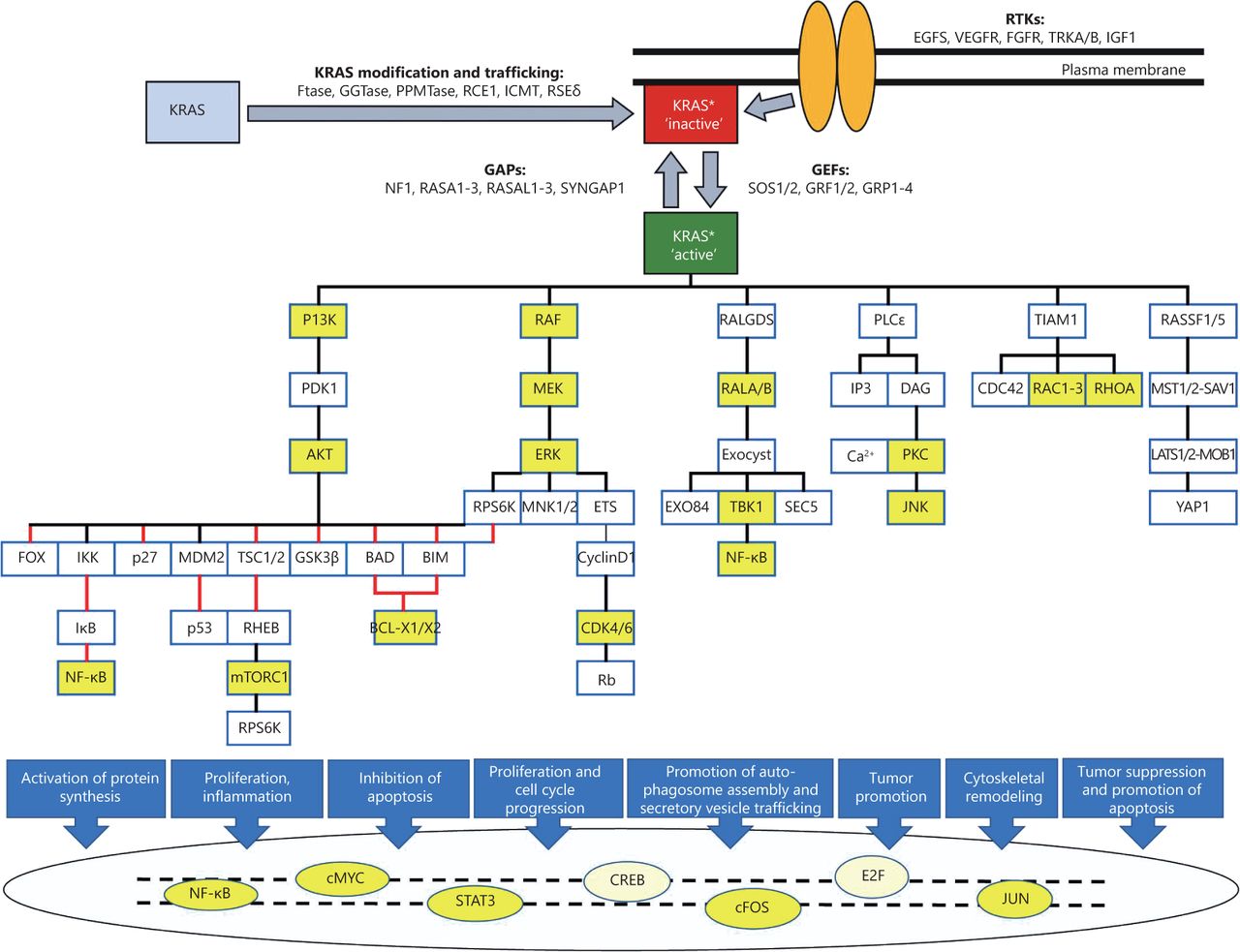
KRAS is a small GDP/GTP-binding protein that transduce extracellular signals into intracellular responses. It cycles between an inactive, GDP-bound (off) state and an active, GTP-bound (on) state. This off/on cycle is tightly regulated by RAS-specific guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs)1. In its active state, KRAS binds and activates various effector proteins function and thus regulate downstream signaling pathways (Figure 1). The conserved GDP/GTP-binding (G) domain of KRAS contains two flexible regions, the switch regions 1 and 2, which provide a functional platform for the interaction with regulators and effectors2-4. The C-terminus of KRAS, which is highly variable among the RAS paralogs, is the site for post-translational modifications and responsible for KRAS anchorage to the plasma membrane5-7.

KRAS signaling pathways. Different upstream RTKs, regulators (GEFs and GAPs), downstream effectors, and transcription factors are presented along with posttranslational modification of newly synthesized KRAS (gray box) to trafficking modified KRAS (red box) and its association with plasma membrane. Stimulatory effects are shown in black lines and inhibitory effects in red lines. The color yellow shows some of the downstream therapeutic targets mentioned in this article. The asterisk * highlights posttranslationally modified KRAS.
Upstream signaling pathways of KRAS are activated by binding of ligands to their transmembrane receptors, mostly receptor tyrosine kinases, and recruitment of docking proteins, such as GRB2, in complex with RAS-specific GEFs, which facilitates KRAS activation (Figure 1)8-10. GTP-bound KRAS further transduces the signal to its downstream effectors and thus activates multiple signaling pathways 11-15. Thereby, KRAS controls various cellular processes, including survival, growth, proliferation, differentiation, and apoptosis 16-18.
With the discovery of the mutational activation of RAS genes in human cancers dating back to the 1960s, extensive studies have been conducted to understand the localization, regulation and signaling of RAS proteins with the ultimate goal of developing anti-RAS drugs for cancer treatment3. Somatic mutations, most frequently identified KRAS4B (oncogenic KRAS or KRASonc) (COSMIC), contribute to robust gain-of-function effects and to various types of cancers as well as leukemia and lymphoma tumors19-22. Due to reduced GTP hydrolysis and resistance to GAPs19,20, KRASonc persist in a constitutive active state and thus, strongly contribute to neoplastic signal transduction23.
Despite intensive efforts on the understanding of the mechanisms of intracellular trafficking, regulation and signaling activity of RAS proteins, specific inhibition of oncogenic RAS has not been clinically established to date3. Among the RAS protein family, KRAS mutations are the most common oncogenic driver in many human cancers4. Additionally, KRASonc is a strong predictive biomarker of resistance to anti-EGFR (Epidermal Growth Factor Receptor) treatment. Therefore, the prevalence of KRAS mutations in a number of human cancers and its inherent resistance to anti-EGFR targeting underscores the clinical relevance of targeting KRASonc in cancer treatment2,24.
Extensive research on different cell lines harboring the KRAS mutation have been conducted, including a pancreatic cancer cell line (PANC-1)25, human colorectal cancer cell lines (DLD-1, HCT-116, and Colo-320 cells)26, non-small cell lung cancer (H441 cells)27, human bronchial epithelial cells (HBEC3KT cells)28, human alveolar basal epithelial cells (A-549 cells)29, human oral squamous cell carcinoma (H157 cells)29, human breast adenocarcinoma cells (MCF-7 and SKBR3-LR cells)30, murine embryonic fibroblasts (MEFs)31, and acute myeloid leukemia cells (NOMO-1)25. According to studies on targeting the KRAS oncogene, therapeutic strategies can be divided into two main categories: 1) small molecule inhibitors, which are synthetically lethal to mutant KRAS or designed to prevent the post-translational processing of KRASonc, upstream pathways, KRASonc/GEF interactions and downstream KRAS* effectors; and 2) anti-KRAS genetic therapies, which interfere with the expression of KRAS or other components of KRASonc-associated signaling pathways.
The complexity of KRAS signaling pathways, in which KRAS protein interacts with many different upstream mediators, downstream effectors, and transcription factors in a nonlinear fashion, has a critical role in the lack of effective treatment32-34. Thus, a better understanding of KRAS interactions with other proteins and transcription factors may provide new opportunities for effective treatment (Figure 1).
In this review, we provide a snapshot view of the rich history of KRAS research by chronologically discussing representative key retrospective discoveries regarding the various therapeutic options for cancers associated with KRAS mutations. In addition to basic original anti-KRASonc therapeutic mechanisms, novel approaches, including inhibition of the embryonic stem cell-like program18, targeting of upstream tyrosine kinases10, stabilization of KRASonc G-quadruplex structures35, inhibition of inflammation36, and targeting of metabolic peculiarities37, for suppression of aberrant KRAS activation in cancers are also explained (Figure 2).

Different therapeutic targets for KRAS driven cancers. The most important of these therapeutic strategies discussed in this article are shown by numbers: (1) Inhibition of transcription by G4 elements. (2) Inhibition of translation through complementary microRNAs. (3) Targeting enzymes posttranslationally modifying KRAS. (4) Targeting KRAS membrane trafficking. (5) Interference with upstream signaling by targeting of receptor tyrosine kinases. (6) Targeting GEFs and RAS activation. (7) Targeting KRAS effectors and downstream signaling pathways. (8) Suppression of synthetic lethal interactions. (9) Targeting inflammatory signaling pathways. (10) Targeting cell cycle progression. (11) Reregulation of metabolic alternations. (12) Reprogramming of stem cell properties. (13) Upregulation of miRs with anti-KRAS activity. Black arrows with blocked red circles are referred to inhibited targets as potential therapeutic approaches.
In addition to KRAS mutations, amplification of wild-type KRAS gene or EGFR mutation leads to the over-expression or over-activation of KRAS, respectively. Some studies have shown that both over-expressed KRAS and KRASonc can be associated with aggressive and metastatic cancer phenotype38,39. Regarding these similarities, some of the targeting strategies discussed in this review may be applied for both KRAS and KRASonc, e.g., inhibition of downstream signaling pathways or inhibition of plasma membrane localization. In contrast, structural differences between KRASonc and KRAS provide distinct therapeutic opportunities40. Some studies, which are referred to in this review, focus on total RAS proteins. Considering that the KRAS mutation represents approximately 90% of identified RAS mutations33, the results of studies on total RAS proteins could certainly be applied to KRAS protein.
Inhibition of KRAS localization
KRAS localization in the plasma membrane is a critical step for its activation and signaling41. Thus, inhibition of KRAS localization provides new insights for cancer treatment. There are three main approaches to prevent KRASonc localization: 1) inhibition of KRASonc post-translational modifications, 2) displacement of KRASonc from the membrane, and 3) impairment of proper KRASonc intracellular trafficking41-43. After translation of KRAS protein, it must undergo a series of post-translational modifications, which facilitate its association with the cell membrane.
Initially, the enzyme farnesyl transferase (FTase) catalyzes the addition of a farnesyl isoprenoid moiety to the thiol group of the terminal cysteine in the CAAX motif of KRAS protein44. CAAX stands for C, a cysteine, A for aliphatic amino acids and X for any amino acid. Next, protease RAS-converting enzyme-1 (RCE-1) cleaves the terminal AAX amino acids, and then the carboxyl group of the cysteine is methylated by isoprenyl-cysteine carboxymethyl transferase-1 (ICMT-1)43,45,46. Multistep post-translational modifications of KRAS protein provide several possible drug targets, including FTase, RCE-1, and ICMT-143,44. Thus, attempts have been made to target KRAS post-translational modifications to inhibit its membrane localization and thus its activation and downstream signaling for the treatment of cancers. Prevention of KRASonc processing to form a stable interaction with the cell membrane is not the only mechanism to reduce the population of KRASonc at the membrane. Displacement of KRASonc from the membrane and the impairment of proper trafficking are the two other strategies47,48. For instance, perturbation of the subcellular distribution of phosphatidylserine leads to a significant reduction of electrostatic interactions between KRASonc and the plasma membrane, resulting in its displacement from the membrane. Another strategy triggering the mislocalization of KRASonc is phosphorylation of S181 in the C-terminal hypervariable region (HVR) of KRASonc, thereby activating the farnesyl-electrostatic switch.
Targeting post-translational modifications of KRASonc to inhibit its plasma membrane localization appeared to be promising in preclinical studies; however, alternative post-translational modifications of KRASonc and disruption of the prenylation of proteins other than KRASonc have led to disappointing results. In spite of the earlier unsuccessful results, continuation of the studies on the disruption of KRASonc plasma membrane localization has led to the development of novel treatment outcomes. For example, RAS binding proteins, such as phosphodiesterase delta subunit (PDEd), have attracted considerable attention as a new target42,49. Prenylation of KRAS increases its hydrophobicity and, thus, reduces its solubility. PDEd facilitates the distribution of RAS family proteins by covering hydrophobic group. Therefore, inhibition of the RAS-PDEd interaction prevents oncogenic RAS (RASonc) activation and signaling and results in an anti-cancer effects on RAS-transformed cells. In recent studies, blockade of the prenyl-binding pocket of PDEd demonstrated promising result42,49. In order to have a view on the progress has been made for disruption of KRASonc plasma membrane localization, studies examining the blockade of KRASonc processing, mislocalization, and trafficking published from 1993 to 2016 are summarized chronologically (Table 1).
Inhibition of RAS plasma membrane localization
Direct inhibition of KRASonc
In response to extracellular stimuli that activate cell surface receptors, RAS protein members mediate the transduction of extracellular signals to intracellular responses. Small GTPases of the RAS family function as molecular switches that cycle between active, GTP-bound and inactive, GDP-bound states66. Activation of upstream signaling pathways results in the recruitment of GEFs, such as SOS1 and SOS2, which facilitate KRAS activation by catalyzing the release of GDP from KRAS67,68. Activated KRAS controls different cellular processes that are also involved in the transformation of normal cells to the malignant phenotype69.
The intrinsic GTPase activity of wild-type KRAS is enhanced by GAPs; however, oncogenic KRAS mutations lead to the impairment of GTP hydrolysis and cause GAP insensitivity and thus constitutive activation of KRASonc70-73. Indeed, inhibition of the constitutively active KRASonc is a conceptually ideal strategy for cancer therapy. Two general mechanisms have been suggested for direct inhibition of RAS proteins, including decreasing the proportion of KRASonc in its GTP state and disrupting the KRASonc-effector interactions. To decrease KRASonc-GTP levels, several approaches have been used, such as the inactivation of KRASonc with small molecules or GTP analogs that facilitate GTP hydrolysis activity, interference with the nucleotide exchange process through disruption of the SOS-KRASonc interaction, subversion of the native nucleotide preference of the KRASonc to favor GDP over GTP, irreversible inhibition of the KRASonc with its covalent modification, inactivation of KRASonc in the GTP state, inhibition of intrinsic nucleotide exchange, and inhibition of nucleotide binding40,67,74.
Activation of downstream effectors, such as RAF kinases, is accomplished through direct interaction of KRASonc with its effectors. Likewise, other approaches in treatment of KRASonc-driven cancers, first generation of RAF kinase inhibitors had limited clinical benefit where the inhibitors found to paradoxically activate ERK pathway through the induction of RAF dimerization in RAS-mutant cancers75. Discovery programs in the development of new RAF inhibitor compounds overcome limitations associated with RAF dimerization. Next generation inhibitors take two approaches to combat RAF dimerization. The first approach is the development of compounds with the equal potency for inhibition of both monomeric and dimeric RAF. The second strategy is the recruitment of ATP binding cleft to disrupt RAF dimerization75. Other than these therapeutic strategies, progress has been made in generating alternative agents to inhibit KRASonc-RAF interaction which is needed to stimulate RAS-dependent oncogenic signaling40,76. Thus, a better understanding of the detailed interactions of KRASonc with RAS binding domains and RAS association domains of its downstream effectors provides alternative opportunities for the inhibition of intermolecular interactions77,78. Table 2 provides a summary of studies examining the direct inhibition of KRAS mutant from 1997 to 2017.
Direct inhibition of KRAS mutant as therapeutic strategy
Direct inhibition of KRASonc probably one of the most important therapeutic strategies, has some drawbacks. Direct targeting of this oncogene is difficult owing to its picomolar affinity for GTP/GDP. Furthermore, the interaction of KRAS with small molecules that facilitate GTP hydrolysis is challenging because the active site is occupied by guanine nucleotides, and there is little space for binding small molecules94. KRAS molecular switching and signaling are accomplished by protein-protein interactions. Inhibition of these interactions requires a detailed understanding of the interacting interfaces and their characteristics. Additionally, the relative featureless topologies of these surfaces and poor drug-like properties of peptides that disrupt protein-protein interactions make the inhibition more challenging73. While targeted therapy against many cancers, such as EGFR-mutated cancers, provides effective responses, no FDA-approved KRASonc-targeted therapy is currently available, and cytotoxic chemotherapy remains the best option for patients with KRASonc-driven cancers. Hopefully, following the earlier failures in the direct inhibition of KRASonc, a new wave of research in recent years has provided promising results. The KRAS oncoprotein has some specific structural features in comparison to wild-type KRAS. Selective targeting of these differences allows direct inhibition of the KRAS mutant without affecting wild type KRAS. For example, recent studies focusing on the KRAS-G12C mutation as a direct inhibition strategy have been showed significant results. In this type of mutation, the thiol group of the cysteine residue located close to the nucleotide-binding pocket, switch I, and switch II, are targeted by different small molecules that result in the inhibition of downstream interactions. Since KRAS-G12C is the most common mutation in lung cancer patients, the translation of this agent to clinical practice would be a significant approach for generating novel anti-KRASonc therapeutics40,67.
RNA interference
The KRAS oncogene activates multiple downstream cellular pathways to drive the progression of cancer1,95. Because of the unsuccessful EGFR targeted therapy for KRASonc-dependent cancers and the difficulty associated with targeting KRASonc directly, a great deal of effort has been applied to target downstream effector pathways. The specific interaction of RAS family proteins with downstream effectors regulates various cellular functions3,77,96,97. Constitutive activation of downstream effector pathways by oncogenic KRAS results in the uncontrolled growth, proliferation, and survival of cancer cells98. It is essential to identify the effector pathways that are required for KRAS-driven carcinogenesis to identify pathways that should be targeted for treatment99.
Two of the best-characterized KRAS effector pathways are the RAF-MEK-ERK and PI3K-AKT-mTOR pathways, which are integral to KRASonc-driven transformation through different signaling cascades100–102. These pathways comprise different kinases, providing multiple nodes for potential therapeutic intervention103,104. Collectively, studies on targeting the RAF-MEK-ERK and PI3K-AKT-mTOR pathways are divided into two categories. The first series of the studies focused on the identification of compounds targeting only one of the downstream signaling pathways, including RAF inhibitors, MEK inhibitors, or PI3K inhibitors (Table 3).
Targeting downstream signaling pathways of RAS as therapeutic strategy
The results of these studies have shown that, due to the interplay between downstream signaling pathways of KRAS, inhibition of one downstream target leads to the overexpression of its interconnected pathways, creating a drug-resistant phenotype. For example, in response to MEK inhibition, PI3K is activated through a negative MEK-epidermal growth factor receptor-PI3K feedback loop32,112. Therefore, novel therapeutic approaches are focusing on the disruption of these multiple nodes, which is only possible through the inhibition of multiple downstream kinases, rather than only one through combination therapy100-104 (Table 4).
Targeting downstream signaling pathways of RAS as combination therapy
According to the valuable results from the combination therapy, extensive studies are moving forward based on multi-targeted therapy for the inhibition of KRASonc downstream signaling pathways. Recently, a large trial investigated the therapeutic effects of the MEK inhibitor selumetinib and docetaxel in comparison to docetaxel alone, producing results in NSCLC patients with the KRAS mutation114. Other results from an ongoing trial show a clinical benefit from combination therapy with an investigational MEK inhibitor known as PD-0325901 and palbociclib, an inhibitor of CDK4/6 (PD-0332991), in patients with KRAS-mutant NSCLC (NCT03170206) and KRAS-mutant PDAC (NCT03454035). In addition, phase II of the other ongoing study on investigational drugs GSK2256098 (focal adhesion kinase inhibitor) and trametinib (MEK inhibitor) was planned to evaluate the antitumor activity of this combination therapy in patients with advanced pancreatic cancer (NCT02428270). BVD-523, an ERK inhibitor, is also currently being tested in combination with nab-paclitaxel plus gemcitabine in a phase Ib trial in patients with metastatic pancreatic cancer (NCT02608229). Another downstream inhibitor is mTOR, a component of the PI3K pathway. The mTOR inhibitor (NCT02329717) PBI-05204 has been tested in patients with stage IV pancreatic cancer. In the other clinical trial, the pan-RAF inhibitor (LXH254) and ERK suppressor (LTT462) are being evaluated as combination therapy for patients with advanced-stage solid tumors with mitogen activated protein kinase (MAPK) alterations, including KRAS-mutant NSCLC (NCT02607813 and NCT02974725). Additionally, phase I/II trials have been initiated to assess the combination therapy of the MEK inhibitor trametinib and the BCL-XL and/or BCL-2 inhibitor navitoclax in patients with KRAS-mutant advanced-stage solid tumors (NCT02079740).
Response evaluation criteria in solid tumors
RNA interference (RNAi) is based on a natural process by which RNA molecules inhibit the generation of protein from DNA115,116. For example, in the search for novel strategies in the treatment of KRASonc-driven cancer, microRNAs (miRs) have received attention for their role in the regulation of gene expression30,117. MiRs are small, single-stranded, highly conserved non-coding RNA molecules that are involved in the control of gene expression118,119. These molecules exert their action by binding to target mRNAs to prevent protein production. The degree and nature of the complementarity between the microRNA and target mRNA determines the gene silencing mechanism that will be employed. Perfect complementarity to the mRNA target leads to its subsequent degradation and transcriptional inhibition, while partial complementarity results in the blockade of translation120. Therefore, this complementarity plays a key role in regulating the target gene of a particular microRNA. For instance, polymorphisms of the let-7 microRNA binding site in the 3' untranslated region of KRAS leads to an impairment of their complementarity and elevated expression of KRAS117,121,122.
The dysregulation of microRNAs and their critical roles in carcinogenesis results from the ability of microRNAs to control the expression of oncogenes and tumor suppressor genes123. For a microRNA with tumor-suppressor activity, its downregulation promotes tumorigenesis, while overexpression of a microRNAs with oncogenic effects leads to cancer development. Mechanisms responsible for the deregulation of miRs in cancers can be classified as genetic and epigenetic alterations that are observed in cancer cells124. Considering KRAS as a proto-oncogene, downregulation of miRNAs that suppress KRAS activation and activation of miRNAs that modulate KRAS expression can lead to cancer development125. Some of the microRNAs directly target KRAS, and some of them suppress KRAS activation through other targets (Table 5). For example, the results of a study showed that KRASonc suppresses mir-200 family expression through its downstream effectors JUN and SP-1119. An alternative RNA therapeutic approach to miRNAs is through the use of small interfering RNAs (siRNAs) and short hairpin RNAs (shRNAs) from the family of non-coding RNAs115 (Table 5). SiRNAs also regulate gene expression through gene silencing with inhibition of gene translation into protein126. Additionally, their similarity in structural characteristics and pharmacokinetic profiles facilitates their use as therapeutics127.
Efficacy of RNA therapeutics on KRAS targeting
Regarding RNA-mediated silencing method, there are two strategies to suppress KRAS oncogenic signaling. First strategy is direct in which KRAS gene expression is reduced by direct binding of RNAi to KRASonc mRNA (Table 5).
Second approach is indirect, which is based on the inhibition of synthetic lethal interactions. Synthetic lethality is a phenomenon through which a genetic alteration leads to cell death only in the presence of another genetic perturbation. Mechanistically, synthetic lethal interactions can involve genes that are functionally connected148. In cancer cells, aside from pathways directly controlled by oncogenes, there are several non-oncogene-targeted pathways, which are involved in the process of transformation149. Thus, oncogenes require additional support from other genes to maintain the oncogenic state26. A major challenge in cancer treatment is the identification of targets that can be inhibited for the selective killing of cancer cells while sparing normal cells. Synthetic lethal interactions between oncogenes and non-oncogenes in cancer cells increase the sensitivity of cancer cells to selective therapeutics in comparison to normal cells150.
The KRAS mutation predisposes cancer cells to additional dependencies on the activity of genes that are not directly regulated by KRAS. This phenomenon can provide an approach for the selective treatment of KRASonc-driven cancers according to the synthetic lethal interactions26,151. The KRAS signaling pathway is complex, so several potential synthetic lethal targets are required for the initiation or maintenance of KRAS mutant tumors. To identify critical nodes in the signaling pathways regulating aberrant KRASonc signaling, RNA silencing technologies could be exploited152. These newly identified synthetic lethal interactions lead to novel therapeutic opportunities. Additionally, small-molecule synthetic lethality screens have resulted in the identification of the selective effect against KRAS mutant cells compared with wild-type cells31,153. It should be noted that inhibition of synthetic lethal interactions is not only accomplished by RNAi, but also with small molecules. A summary of researches on the screening of synthetic lethal interactions with KRASonc through RNA silencing methods or small molecules is provided in Table 6.
Inhibition of synthetic lethal interactions of KRAS as a therapeutic strategy
Antisense oligonucleotides directed against KRASonc have indicated a therapeutic benefit in laboratory studies, opens up multiple effective possibilities for suppressing KRAS activity, and preventing the feedback response and drug resistance while facilitating combination therapy99. Despite the tremendous potential of RNA-based therapies, the successful application of this technology is currently limited. RNAs are inherently unstable, and therefore there is lack of efficient delivery of sufficient amounts to the target tissue. Additionally, toxicity due to off-target effects and the induction of immune system responses also represent difficulties related to this approach115,158,159.
Targeting the immune system
Cancer immunotherapy for patients carrying the KRAS mutation has become a clinical oncology reality. KRAS-G12D knockdown cells show increased production of interleukin 18 by the host immune system, leading to a dramatic reversion of the transformed phenotype and reduction of the proliferation rate of cancer cells129. Increases in NK cells and antibody-dependent cell-mediated toxicity after combination therapy with lenalidomide and cetuximab lead to increases in circulating na and central memory T cells in patients with KRAS-mutant colorectal cancer160. The KRAS mutation induces increased expression of programmed cell death 1 ligand 1 (PD-L1)161. According to these findings, Ebert et al. showed that anti-PD-L1 antibodies significantly reduced tumor size in MEK inhibitor-treated mice with the KRAS mutation. Thus, it seems that the combination of immunotherapy and anti-proliferative agents, such as MEK inhibitors, provides higher anti-tumor activity162. Genetic alterations are specific to cancer cells and are not present in normal cells; thus, treatments that specifically target the protein product of these genetic aberrations may provide a clinical benefit in the absence of normal cell toxicities. Although mutant KRAS proteins themselves are not strongly immunogenic, efforts are underway to enhance the ability of the immune system to recognize KRAS mutant peptides as neo-epitopes. For example, specific immunogenic mutations could help to recognize KRAS mutant variant peptides of the most frequent KRAS mutations, such as G12V and G12D, by specific T cell receptors163. In this way, to develop more effective personalized immuno-therapy for patients with the KRAS mutation, Rosenbergs team isolated tumor-infiltrating lymphocytes (TILs) with the ability to specifically target the KRAS mutation. The findings of that study, which were presented in December 2016, introduced, for the first time, a novel immunotherapy-based strategy, called adoptive T cell transfer immunotherapy. These results validated the possibility of using personalized T cell receptor gene therapy against multiple types of cancer expressing this common mutation or other types of KRAS mutations163.
Thus, the purpose of recent studies has been the identification of immune-editing of T cells during tumor development, as well as the determination of their potential applications for tumor-specific immunotherapy164.
According to the brilliant results from immunotherapy, treatments focused on altering the immune system for patients suffering from KRASonc-driven cancers have been intensively investigated in recent years, with new achievements. In one study, the efficacy of immune checkpoint inhibitors among NSCLCL patients was found to correlate with the KRAS mutation as a molecular smoking signature165. Other evidence indicates that the co-mutation of TP53 and KRAS in lung adenocarcinoma can be exploited as a potential predictive marker for effective immune checkpoint blockade immunotherapy164. Clinical trials have also been initiated for the KRAS-G12D-specific cancer vaccine TG01/ GM-CSF either alone or combined with gemcitabine. The initial results of these trials have shown an induction of the immune responses in response to TG01/GM-CSF plus gemcitabine combination therapy166. A study to evaluate the efficacy and safety of cobimetinib plus atezolizumab and atezolizumab monotherapy versus regorafenib in participants with metastatic colorectal adenocarcinoma is currently ongoing as a phase III trial (NCT02788279). The initial findings suggest that this therapeutic strategy is helpful in improving the immune response. One trial examining the combination therapy of a newer CDK4/6 inhibitor, abemaciclib, with the immune checkpoint inhibitor pembrolizumab is currently ongoing in NSCLC patients with the KRAS mutation167. New achievements have been observed in these studies against human cancers (Table 7), represent the need for further studies to enhance immunotherapeutic efficacy in some patients.
Studies on immune system targeting RAS-driven cancers
Other approaches
Despite important strides made in the development of targeted therapy for KRASonc-mediated cancers, no therapeutic approaches are clinically available. In recent years, a deeper understanding of the critical parameters involved in the promotion of KRASonc-driven tumorigenesis has been considered for the development of new therapeutic options. In this part of the article, we review these new achievements and discuss multiple lines of evidence of novel key pathways that are recognized to interact with other previously identified KRAS-regulated survival pathways to transduce signals of carcinogenesis. The data suggest that co-targeting of these newly and previously recognized KRASonc-regulated pathways has significant clinical potential.
Inhibition of stem cell program
Cancer stem cells (CSCs) are defined as tumor-initiating cells with self-renewal capacity. They are considered to be responsible for cancer initiation, progression, metastasis, drug resistance, and treatment relapse168. The KRAS mutation has been shown to preferentially alter the profile of gene expression to induce embryonic stem cell-like features169. For example, the expression of some genes is known to be upregulated in the presence of the KRAS mutation, including fibroblast growth factor receptor 1 (FGFR1), which plays a common role in both embryonic and cancer development, LCK, the transcriptional silencing of which is required for embryonic stem cell differentiation, and the induced-pluripotency factor SOX2, which reprograms differentiated cells to pluripotency. In contrast, KLF4 expression was suppressed in KRAS mutant colon cancer cells, which is consistent with its induction of multiple cell lineage differentiation in the intestine18. Additionally, a KRAS-centric mechanism would apply in the context of epidermal-mesenchymal transition (EMT) to generate CSCs through the WNT pathway170.
Other results have indicated that oncogenic KRAS activation in the genetic background of loss-of-function of adenomatous polyposis coli (APC) results in enhanced CSC activation by increasing both intracellular stabilization of ݭcatenin and the MAPK pathway171,172. Furthermore, endodermal progenitors expressing KRAS-G12V do not differentiate upon retinoic acid treatment and continue to proliferate and maintain stem cell characteristics173. Several studies have described the KRAS mutation as a driver of stem cell-like properties of cancer cells. Thus, inhibition of multiple key pathways involved in embryonic stem cell signaling represents a novel therapeutic strategy. Le Rolle et al.18 showed that inhibition of KRAS mutant colon tumors with miR145, an epigenetic regulator and an embryonic stem cell inhibitor, suppressed their malignant growth. Data suggest that salinomycin, the most potent cancer stem cell inhibitor with potential efficacy in human cancers, specifically disrupts KRASonc nanoscale membrane organization, effectively reducing effector recruitment to KRASonc, which then compromised at least MAPK signaling and proliferation170. Ophiobolin A, another candidate CSC drug, has been found to possess higher potency than salinomycin and exert its KRAS4B-specific activity through the inactivation of calmodulin170.
Based on the role of the KRASonc in stemness, a-Mangostin-encapsulated PLGA [poly (D, L-lactic-co-glycolic acid)] nanoparticles show inhibitory effects on carcinogenesis in transgenic mice carrying the KRAS mutant allele through the downregulation of pluripotency maintenance factors (c-MYC, NANOG and OCT4) and stem cell markers (CD24 and CD133)174. Overall, these data suggest that targeting multiple signaling pathways of cancer stem cell activation induced by the KRAS mutation could be an attractive therapeutic approach.
Targeting receptor tyrosine kinases (RTKs)
A growing body of evidence suggests that the KRAS mutation may serve as a predictive resistance marker to guide the use of anti-EGFR therapy. Multiple studies have demonstrated that patients with mutations in KRAS do not appear to experience a clinical benefit from anti-EGFR monoclonal antibody treatment175. In cancers with KRAS mutations, part of the cell survival and proliferation pathways could still be due to the activation of upstream RTKs other than EGFRs. Therefore, another possible approach to target tumors with KRAS mutations is through the inhibition of such critical RTKs that contribute to the enhanced prosurvival. The type 1 insulin-like growth factor receptor is a promising target in different types of cancers, including colon cancer176. The PI3K signaling pathway is a common downstream effector of both IGF-1R and KRAS. Thus, blockade of IGF-1R using different monoclonal antibodies or tyrosine kinase inhibitors is theoretically relevant for the treatment of patients with KRASonc-driven cancers8. Although patients with the KRAS mutation show resistance to EGFR-targeted therapy, preclinical data have indicated that combination therapy with IGF-1R and EGFR kinase inhibitors results in synergistic growth inhibition in colorectal cancer cell lines9. Hurwitz et al.10 showed a clinical benefit following the treatment of patients with bevacizumab as an anti- vascular endothelial growth factor (VEGF) therapy. Data have also shown that, unlike anti-EGFR therapy, anti-VEGF therapy functions independently of the KRAS mutation status, revealing even greater clinical significance.
Stabilization of the G-quadraplex
G-quadruplexes (G4) are special secondary structures containing runs of guanines separated by other bases177. The localization of G4 in the human genome was found to be non-random, indicating their important role in the regulation of functional regions. Significantly, G4 are more frequent in oncogenes or regulatory genes than in house-keeping or tumor suppressor genes. Their higher distribution in the promotors of oncogenes suggests a possible involvement of G4 in cancer178. Genome-wide analysis of human cells has revealed the role of these structures is gene-silencing through the inhibition of replication, transcription, and translation35. Therefore, the stabilization of guanine-rich regions located in the oncogene promoters represents a highly valuable new molecular target for the development of novel anti-cancer therapeutics177. It is now evident that the core promoter region of KRAS contains silencing G4 elements179. G-to-T knockout mutations in the G4-forming regions of the KRAS promoter were found to disrupt or abrogate G4 formation. In addition, stabilization of the KRAS promoter by the cationic porphyrin TMPyP4 leads to a significant decrease in KRAS expression180. The interaction of G4 of the KRAS promoter with natural polyphenols, such as ellagic acid and curcumin, has also been confirmed by UV-vis spectroscopy. Significantly, the melting temperature of the G-quadruplex is increased, indicating its stabilization upon interaction with polyphenol ligands35.
Inhibition of inflammation
KRAS-driven tumorigenesis is tightly connected with tumor-promoting inflammation, which increasingly represents another promising therapeutic strategy181. According to recent clinical data indicating the role of inflammation in the carcinogenesis related to the KRAS mutation, targeting inflammatory signaling pathways seems to be an essential component of therapy for tumors with KRAS mutations182. Different cellular pathways, which are modulated by KRAS and induce inflammation, include JAK/STAT, NF-κB, MAPK, and immune checkpoint signaling pathways36. For example, the KRAS mutation contributes persistent pancreatitis induced by cerulein. In this situation, suppression of inflammation by deletion of IKK-ݠand inhibition of NF-κB activity interferes with dysplasia. In contrast, overexpression of IKK-ݠcooperates with the KRAS mutant allele to promote oncogenesis183.
A different study indicated that while persistent KRAS activation drives the secretion of STAT3 pathway mediators, activation of STAT3 results in the amplification of KRASonc carcinogenesis through the upregulation of anti-apoptotic and pro-proliferative proteins184. Co-administration of azoxymethane (AOM) and dextran sodium sulfate (DSS), respectively, as carcinogenic and inflammatory agents, results in a significant decrease in the latency of KRASonc-driven tumor formation185. Given the presence of inflammatory stimuli in a KRAS mutation background as positive feedback promoting KRASonc-associated carcinogenesis, targeting each of the mentioned signaling pathways would likely lead to the development of a mechanism for disease control.
Targeting metabolic pathways
Metabolic reprogramming of cancer cells due to oncogenic mutations is critical for cell growth and survival. Data show that the KRAS oncoprotein confers metabolic robustness for the acquisition of cellular metabolism networks to convert carbon sources into biomass186. The metabolic features of KRASonc-driven cancers can be explained through the reprogramming of glucose, amino acids, and lipid metabolisms37. Cancer cells harboring KRASonc promote the glycolytic switch, glucose uptake, increased channeling of glucose-derived metabolites into the tricarboxylic acid cycle, and activation of glucose-dependent biosynthetic pathways187. For example, it has been reported that the KRAS mutation increases the expression of glucose transporter-1 (GLUT1) and several rate-limiting glycolytic enzymes188. Interestingly, the induction of metabolic changes is dependent on the content of the KRAS mutant allele of cancer cells. Thus, glycolytic gene expression was markedly enhanced in KRAS-G12D/G12D relative to heterozygous lung tumor cells187. One mechanism by which KRASonc aberrantly regulates metabolic networks is through the reprogramming lipid metabolism by the promotion of cellular uptake, retention, accumulation, synthesis, and oxidation of fatty acids. For instance, lung cancer cells carrying the KRAS mutation are highly dependent on the activity of acyl-coenzyme A synthetase long-chain family member 3 (ACSL3)28,189. Mutated KRAS promotes lipogenesis through the induction of fatty acid synthase, leading to lipid signatures of human lung cancer cell lines189. Other results have shown that the RAS mutation leads to the reprogramming of de novo lipogenesis of cancer cells by scavenging serum fatty acids190. Emerging evidence from different research groups indicates that KRAS mutations are associated with changes in amino acid metabolism191. Reprogramming of glutamine metabolism in KRASonc-driven cancers is the most important alteration in amino acid metabolism. While most cells utilize glutamate dehydrogenase 1 for conversion of glutamate into a-ketoglutarate, cancer cells carrying the KRAS mutation convert glutamate to aspartate191. The increased requirement for branched-chain amino acids (BCAAs) is a very early phenomenon during tumor development, similar to some types of KRASonc-driven cancers192. As mitochondrial activity is required for metabolic changes in cancer cells, autophagy as a mechanism for the elimination of defective mitochondria is crucial for tumor growth. Loss of essential autophagy genes in KRASonc-driven cancer impairs effective mitochondrial function and suppresses tumor progression, emphasizing the role of autophagy in the intracellular nutrient supply193. These reports indicate that the KRAS mutation creates unique metabolic dependencies that could be exploited for anti-cancer therapy.
Targeted RNA replacement
Tetra hymena group I intron-based trans-splicing ribozyme is specific therapeutic tool with ability to discriminate the target RNA resulting in specific and high-fidelity cleavage reaction of its target194. Moreover, ribozymes can specifically transfer the therapeutic gene into cancer cells expressing target RNA. This specific trans-splicing reaction with the ability of discrimination target RNA from non-target one, even with a single nucleotide difference, makes it as an attractive novel treatment strategy for KRAS point mutations. Regarding KRAS-G12V mutation as one of the most prevalent point mutation, Tetra hymena group I intron-based trans-splicing ribozyme designed for selective cleavage of KRAS-G12V transcript195. An accurate and specific intracellular trans-splicing reaction of the designed ribozyme systems with the KRAS-G12V target RNA, leads to efficient reduction of transcript level. Except that replacement of RNA, concurrent induction of suicide gene activity resulting in cytotoxicity and effective retardation of cancer cells harboring KRAS mutation196. Moreover, trans-splicing and therapeutic anti-cancer gene activity was selectively and efficiently induced only in KRAS-mutant cancer cells without targeting of cells expressing wild-type KRAS195.
Oncogene-induced senescence
Oncogene-induced cellular senescence (OIS) is a complex mechanism of tumor suppression which is thought to be triggered by aberrant activation of oncogenic signaling197. Undisputed role of RASonc in different human cancers, necessitate studies on the RASonc-induced senescence as an alternative treatment strategy. Senescence is not a simple mechanism triggered by only linear series of events and multiple components are required to establish a senescence response. Accordingly, detailed molecular mechanisms underlying OIS should be completely understood to provide adequate mechanistic insight for implementation of RAS aberrant oncogenic signaling against themselves as a potential anti-cancer strategy198.
Basically, there are three pathways which are recruited by KRASonc to induce senescence which are also interconnected. The first pathway is transcriptional repression of pro-proliferative genes like E2F target genes. In addition to the transcriptional repression, a second pathway that is believed to mediate KRASonc-induced senescence is the DNA damage pathway. Oncogene activation induces aberrant DNA replication events, leading to replication stress and subsequent DNA damage198. Consequently, DNA damage and accumulation of proteins involved in DNA damage response, like ATM and CHK2 results in senescence induced by oncogene activation. Finally, a third pathway, which is essential for senescence and recruited under RAS activation is senescence-associated secretory phenotype (SASP). Studies have recognized that SASP mediates RASonc-induced senescence, through the secretion of specific proteins like C/EBPݠtranscription factor199. Notably, the neurofibromatosis type 1 (NF1), encoding a RAS-specific GAP, has been implicated in OIS200. In this context, suppression of Ras and/or PI3K are sufficient to induce senescence, and these events on their own can activate the known downstream mediators of the senescence response (Rb and p53) through a variety of mechanisms200 (Figure 1). Moreover, in BRAF-driven melanomagenesis, loss of NF1 cooperates with RAF mutations by increasing PI3K/AKT signaling and preventing entry into OIS201,202. While the significant role of the oncogenic RAS in human cancers has been proved for many years, a better understanding of the molecular basis of RASonc-mediated senescence, allows the delineation of new therapeutic approaches surprisingly aimed at engagement of oncogenic signaling against oncogenic signaling.
Conclusions
More than 30 years of intensive research and tens of thousands of published studies have provided valuable insights into the biology, biochemistry and biophysics of RAS family proteins. Signal transduction of RAS (most notably KRAS) is regulated by three classes of canonical interacting partners, including regulators that control activation of the GTPase cycle (by GEFs), its inactivation (by GAPs), and a wide spectrum of effectors (e.g., RAF kinase and PI3 kinase) that initiate signaling cascades downstream of RAS and RAS-like proteins. We have gained deep knowledge about their membrane trafficking, structure-function relationship, mechanisms of GDP/GTP binding and accelerated nucleotide exchange by GEFs, intrinsic and GAP-stimulated GTP hydrolysis, interaction with effectors and activation of diverse signaling pathways. However, these studies have their own eligibility confinement: cell-free investigations have been predominantly carried out in the absence of lipid membrane, using defined domains rather than full-length proteins, and cell-based studies have mostly been performed via the heterologous expression of tagged genes and their variants in methodologically congenial cell lines. As the omics era is coming to an end and research has decelerated, many new movements have emerged, especially due to the accessibility of new technologies. Several novel mechanisms have been uncovered that have extended our understanding of the role of protein-protein/protein-lipid interactions and various types of post-translational modifications in the modulation of RAS protein activity. Another issue is the activation mechanism of regulators and effectors. Notably, the identification of additional components of the RAS interaction networks is a critical step towards understanding both the relationship between RAS proteins and the selective activation of respective effectors, as well as the molecular signatures required for the spatiotemporal integration and activation of GEFs and GAPs. The identification and functional reconstitution of specific interaction networks by using appropriate liposomes and full-length effector proteins may eventually provide fundamental insights into the functional characterization of multiprotein complexes of RAS and the complete identification of regulatory mechanisms. In this context, an interesting issue, which is increasingly appreciated, is a RAS-membrane interaction that appears to generate RAS isoform specificity with respect to regulator and effector interactions. Currently, it has become more evident that an increasing number of additional RAS binding partners are critical in modulating and integrating RAS in various signaling networks at biological membranes. This phenomenon is likely achieved by scaffold proteins, including CAM, GAL1, GAL3, IQGAP1, NCL, NPM1, SHOC2, SPRY, SPRED1 and GAB1, which may modulate isoform specificity at specific sites of the cell. However, the roles of these additional RAS interaction proteins as novel modulators of RAS signaling remain unclear. Hence, elucidation of the RAS signal transduction requires not only RAS-effector interactions but also additional structures and the interplay of multi-protein complexes. Keeping this in mind, accumulating evidence supports a role for cell type-dependent RAS paralog functions that should prompt future efforts to examine the respective pathways in a more context-specific manner. Excluding driver mutations, passenger mutations accumulate and frequently escape natural negative selection, resulting in several oncological outcomes203. In parallel with standard tumor profiling methods, high-throughput technologies, such as next-generation sequencing, have been employed to shift the treatment paradigms. Thus, further characterization of the heterogeneous identity of patient tumor tissue exploring all specific molecular aberrations along with the specific KRAS mutation, seems to be critical for an effective therapy204,205. Such efforts could lead to the identification of disease-specific therapeutic opportunities. The other novel technology is phosphoprotein analysis through kinome profiling, which provides evidence of signaling pathways that are activated in a patients tumor206.
The authors of this review article conclude that translating our knowledge of different treatment frameworks to the clinic via targeted therapy of the KRASonc and personalized immune-therapy may be the best strategies to dramatically improve patient outcomes. In summary, we are at the beginning of a new series of attempts to treat KRASonc-driven cancers by directly targeting the protein or through personalized targeted therapy with high-throughput or immunotherapy-based strategies. This new wave of personalized studies provide hope for thousands of patients suffering from KRASonc-driven cancers.
Acknowledgments
Authors are thankful to Dr. Seyed Ali Jafari (Mashhad University of Medical Sciences) for insightful comments, and to Dr. Saeideh Nakhaei-Rad for her valuable suggestions. We thank American Journal Experts and Ms. Diana Inanlou for language edition of the manuscript. M.R.A. was supported by the European Network on Noonan Syndrome and Related Disorders (NSEuroNet, Grant No. 01GM1602B), and the German Federal Ministry of Education and Research (BMBF): German Network of RASopathy Research (GeNeRARe, Grant No. 01GM1519D & 01GM1902C).
Footnotes
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received January 18, 2019.
- Accepted June 10, 2019.
- Copyright: © 2019, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.
- 13.
- 14.
- 15.↵
- 16.↵
- 17.
- 18.↵
- 19.↵
- 20.↵
- 21.
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.
- 51.
- 52.
- 53.
- 54.
- 55.
- 56.
- 57.
- 58.
- 59.
- 60.
- 61.
- 62.
- 63.
- 64.
- 65.
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.
- 72.
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.
- 80.
- 81.
- 82.
- 83.
- 84.
- 85.
- 86.
- 87.
- 88.
- 89.
- 90.
- 91.
- 92.
- 93.
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.
- 102.↵
- 103.↵
- 104.↵
- 105.
- 106.
- 107.
- 108.
- 109.
- 110.
- 111.
- 112.↵
- 113.
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.
- 129.↵
- 130.
- 131.
- 132.
- 133.
- 134.
- 135.
- 136.
- 137.
- 138.
- 139.
- 140.
- 141.
- 142.
- 143.
- 144.
- 145.
- 146.
- 147.
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.
- 155.
- 156.
- 157.
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.↵
- 174.↵
- 175.↵
- 176.↵
- 177.↵
- 178.↵
- 179.↵
- 180.↵
- 181.↵
- 182.↵
- 183.↵
- 184.↵
- 185.↵
- 186.↵
- 187.↵
- 188.↵
- 189.↵
- 190.↵
- 191.↵
- 192.↵
- 193.↵
- 194.↵
- 195.↵
- 196.↵
- 197.↵
- 198.↵
- 199.↵
- 200.↵
- 201.↵
- 202.↵
- 203.↵
- 204.↵
- 205.↵
- 206.↵
In this issue






{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.