

Abstract
Objective: To examine the effect of pSer9-GSK-3β on breast cancer and to determine whether the underlying metabolic and immunological mechanism is associated with ROS/eIF2B and natural killer (NK) cells.
Methods: We employed TWS119 to inactivate GSK-3β by phosphorylating Ser9 and explored its effect on breast cancer and NK cells. The expression of GSK-3β, natural killer group 2 member D (NKG2D) ligands, eIF2B was quantified by PCR and Western blot. We measured intracellular reactive oxygen species (ROS) and mitochondrial ROS using DCFH-DA and MitoSOXTM probe, respectively, and conducted quantitative analysis of cellular respiration on 4T1 cells with mitochondrial respiratory chain complex I/III kits.
Results: Our investigation revealed that TWS119 downregulated NKG2D ligands (H60a and Rae1), suppressed the cytotoxicity of NK cells, and promoted the migration of 4T1 murine breast cancer cells. Nevertheless, LY290042, which attenuates p-GSK-3β formation by inhibiting the PI3K/Akt pathway, reversed these effects. We also found that higher expression of pSer9-GSK-3β induced higher levels of ROS, and observed that abnormality of mitochondrial respiratory chain complex I/III function induced the dysfunction of GSK-3β-induced electron transport chain, naturally disturbing the ROS level. In addition, the expression of NOX3 and NOX4 was significantly up-regulated, which affected the generation of ROS and associated with the metastasis of breast cancer. Furthermore, we found that the expression of pSer535-eIF2B promoted the expression of NKG2D ligands (Mult-1 and Rae1) following by expression of pSer9-GSK-3β and generation of ROS.
Conclusions: The PI3K/Akt/GSK-3β/ROS/eIF2B pathway could regulate NK cell activity and sensitivity of tumor cells to NK cells, which resulted in breast cancer growth and lung metastasis. Thus, GSK-3β is a promising target of anti-tumor therapy.
keywords
Introduction
More than 522,000 people die from breast cancer every year, and it still remains the leading cause of cancer-related deaths in women1. Glycogen synthase kinase-3 (GSK-3), a serine/threonine protein kinase, has two main subtypes: GSK-3α and GSK-3β, which have 98% similarity in the catalytic zone with slight difference at the N- and C-termini2. While GSK-3α is mainly involved in the process of glycogen metabolism, the biological function of GSK-3β is more complicated and more intensely correlated with cancer; it is over-expressed in various cancers, including breast cancer. The activity of GSK-3β is regulated by phosphorylation/dephosphorylation at different sites; GSK-3β can be inactivated when it is phosphorylated at Ser9 by diverse signaling pathways3. Akt, protein kinase A/C (PKA/PKC), p70 ribosomal S6 kinase (p70S6K), p90 ribosomal S6 kinase (p90Rsk)4-6, and protein phosphatases such as PP1 and PP2A can inactive GSK-3β by phosphorylating it at Ser97. Phosphorylation of GSK-3β at Tyr216 by kinases, such as Fyn and proline-rich tyrosine kinase 2 (PYK2), and autophosphorylation can activate GSK-3β8,9.
GSK-3β has previously been considered a potential tumor suppressor due to its ability to be phosphorylated and to target oncogenic molecules, including c-Jun, c-Myc, cyclin D1, and β-catenin10. Moreover, recent reports have also supported that GSK-3β is a therapeutic target in cancer owing to its negative regulation of proliferation and survival of cancer cells11-15. It has previously been reported that GSK-3β is associated with breast cancer drug resistance after exposure to chemotherapeutic drugs, such as doxorubicin, anthracycline, and endocrine drug tamoxifen10,16. Studies have shown that the activated GSK-3β acted as a tumor suppressor by negatively regulating epithelial-mesenchymal transition and cancer cell cycle by regulation of Mcl-1, Snail degradation17-19. In addition, GSK-3β inactivates EZH2, a histone methyltransferase, which plays an important role in tumorigenesis, and thus, suppresses breast cancer cell growth and migration20. Despite pSer9-GSK-3β being reported as a biomarker of poor clinical prognosis of breast cancer21,22, the role of GSK-3β in breast cancer remains controversial with its role in immunoregulation for breast cancer not well clarified.
Due to these abilities to spontaneously kill targeted cells without any prior sensitization or MHC restriction, natural killer (NK) cells play a significant role in infection, hematopoietic stem cell (HSC) transplantation, autoimmunity, as well as tumor immune surveillance23. NK cells exhibit the cytotoxicity against a variety of “stressed” cells mainly by receptor-ligand specific recognition and binding24, and a series of cytokines released by NK cells also function to kill target cells25. The natural killer group 2 member D (NKG2D) is the most important activating receptor with multiple ligands (NKG2DLs). The MHC class I-related chain (MIC) A/B and the retinoic acid transcript-1/UL-16 binding proteins (RAET1/ULBP) 1-6, play a decisive role in anti-tumor immune surveillance26. Similarly, murine NKG2D binds to retinoic acid early inducible 1 (Rae1) and H60 and ULBP-like transcript 1(Mult-1)27. NKG2D-NKG2DLs signals via the adaptor molecule DAP10 mediated by phosphoinositide 3-kinase (PI3K) and DAP10 and 12 in mice, and induces degranulation and/or cytokine production in cytotoxic effector cells28.
Previous studies have reported that PI3K/Akt/mTOR and reactive oxygen species (ROS) induce M2 macrophage polarization29, therefore, we studied the effect of PI3K/Akt/GSK-3β and ROS on NK cells. ROS are produced in tumor cells mainly by NADPH oxidases of the NOX family30 and mitochondrial respiratory chain complex (MRCC)31,32, accompanied with a series of changes, including the opening of mitochondrial permeability transition pore33. ROS signaling contributes to proliferation and survival of various tumors, and overproduction of ROS promotes cancer development mainly by inducing genomic instability34. Previous studies have shown that ROS could be a potential target of anticancer therapy35,36. ROS could reduce the cytotoxicity of NK cells37 and reverse the immune suppression. In addition, inhibiting ROS could upregulate the NKG2D/MICA38. Eukaryotic initiation factor 2B (eIF2B), an important rate-limiting protein, catalyzes the guanine nucleotide exchange (GNE) reaction, dissociates GDP from eIF2α-GDP, and releases GTP. The eukaryotic translational initiation factor 2 (eIF2), composed of α-, β-, and γ-subunits, plays a critical role during the first step of protein synthesis. In the presence of GTP, eIF2 interacts with initiator Met-tRNA, delivers it to 40S ribosomes, and forms the 43S pre-initiation complex39. Once the conserved Ser51 of eIF2α in the trimeric complex is phosphorylated, the nucleotide exchange activity of eIF2B is inhibited due to its enhanced interaction with eIF2α40. Previous studies have demonstrated that activated GSK-3β inhibits ROS41, and that ROS induces the phosphorylation of eIF2α42, which results in dysfunction of eIF2B, and consequently, alters the expression of target proteins. Therefore, the aim of our study was to explore the effect of this relationship on NK cells and to elucidate whether inactivated GSK-3β downregulates the expression of NKG2DLs via ROS and eIF2B.
Materials and methods
Material and reagents
Cell culture materials were obtained from Corning (Shanghai, China). A ROS assay kit, Wortmannin, N-acetyl cysteine (NAC), and H2O2 were purchased from Beyotime Biotechnology (Shanghai, China). TWS119 was purchased from Selleck (Texas, U.S.A.). LY294002 was purchased from Promega (Madison, U.S.A.). Salubrinal was obtained from MCE China. CD107a (LAMP-1) ELISA kit were purchased from ElabScience (Wuhan, China), whereas, PGE2 and IFN-γ ELISA kits was purchased from Mlbio (Shanghai, China), and mouse RPS19 ELISA kits were purchased from SaiDongnan (Tianjin, China). Antibodies were purchased as follows: NKG2D and ULBP1 or Mult-1 (Proteintech, America), GSK-3β (Boster, Wuhan, China), pSer9-GSK-3β (Santa Cruz, CA, USA), Rae1 (Santa Cruz), H60 (R&D system, Minnesota, USA), pSer535-eIF2B (Eno Gene, Nanjing, China), and β-actin (Santa Cruz).
Mice
Female BALB/C mice (6 to 8 weeks old) were purchased from the Institute of Experimental Animals of the Chinese Academy of Medical Sciences, and housed in a standard polypropylene cage containing sterile bedding under controlled temperature (23 ± 2 °C), humidity (50% ± 5%), and light (10 h of light and 14 h of dark). All experimental procedures were approved by the medical ethics committee, Nankai University (No.NK23508677EA2018). Transplanted tumor mice model was established by subcutaneously injecting 1 × 106 4T1 cells near the second right mammary fat pad. Seven days later, 12 mice were divided randomly into different 3 groups (4 mice per group) and intraperitoneally injected with different drugs: Vehicle (100 μL physiological saline), TWS119 (15 mg/kg), and LY294002 (5 mg/kg), all once a day for seven days. Tumor growth was measured by a caliper every three days and tumor volume was calculated according to the formula V = (L * W2) * 0.5, where L and W refer to mid-axis length and width, respectively. The mice were sacrificed at the 28th day, eyeball blood was collected for ELISA, tumors were weighed by electronic balance, and partially stored at –70 °C, and the rest was fixed in formaldehyde solution. Lungs were stained with India ink to detect metastasis nodules according to the following protocol: isolate the trachea and inject 15% India ink into the lungs through the trachea until the lungs stop dilating, ligate the trachea to prevent the backflow of India ink, wash the lungs with Fekete’s solution (70 mL 75% alcohol + 10 mL methanol + 5 mL glacial acetic acid + 15 mL distilled water), and analyze images to count the tumor nodules.
Cell culture
Murine breast cancer cell line, 4T1, and murine NK cell line, KY-1, were purchased from ATCC. Primary NK cells were obtained from fresh spleens of BALB/C mice. The mice spleen NK cell isolation optimizing system was purchased from Tianjin Haoyanghuake Biological Product Co. Ltd. 4T1 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution (100 ×) in a humidified atmosphere containing 5% CO2 at 37 °C. Murine primary NK cells were isolated from fresh spleens by TBD Science Mouse NK Cell Separation Kit (TBD science, Tianjin, China). In brief, spleen was ground into single cell homogenate, and after adding separation reagents, NK cells were purified. Purified primary NK cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin solution (100 ×), 200 U/mL interleukin 2 (IL-2; PeproTech, Beijing, China) and 10 μM β-mercaptoethanol in a humidified atmosphere containing 5% CO2 at 37 °C. The drugs used in the study were as below: TWS119 (0–10 μmol/L, 24 h), LY294002 (30 μmol/L, 12–16 h), H2O2 (250 μmol/L, 24 h), and NAC (5 mmol/L, 24 h), and the supernatant of 4T1 cells was added to medium according to the ratio of 2:3 to culture NK cells.
ELISA assay
Eyeball blood was collected from mice, incubated at 37 °C for 30 min, then centrifuged at 3,000 rpm for 10 min, and the serum was transferred to a new micro-centrifuge tube. Supernatants were harvested after NK cells were incubated with 4T1 cells. Both the serum and the supernatants were prepared to analyze the concentration of related factors according to manufacturing instructions with mouse CD107a (LAMP-1), Prostaglandin E2 (PGE2), IFN-γ ELISA kits. The total protein from 4T1 cells was extracted and analyzed by the Mouse RPS19 ELISA kit. All the assays were performed in triplicates.
RT-PCR analysis and real time-PCR
Total RNA (1 μg) was reversed transcribed into cDNA using EasyScript® First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). RT-PCR was performed with a MyGeneTM Series Peltier Thermal Cycler, PCR products were electrophoresed on 1.5% agarose gel and visualized by GelRed staining, and photographs were taken using a Gel DocTM XR+ (BIO-RAD, USA). The real time-PCR reaction was carried out with TransScript top green real time-PCR supermix (TransGen, Beijing, China), and the Bio-Rad CFX Manager system (Bio-Rad, USA). The primers are described in Table 1.
Primer sequences
Western blot analysis
4T1 cells or tumor tissue homogenate were solubilized in RIPA lysis buffer with 1% phosphatase inhibitors and 2% protease inhibitor on ice and cell lysates were isolated by centrifugation at 12,000 rpm for 20 min. Then, the supernatant was transferred to a new micro-centrifuge tube. Protein concentrations were determined using BCA protein assay kit (Beyotime, Shanghai, China), and the proteins were mixed with 5 × loading buffer (protein: 5 × loading buffer = 4:1) and boiled at 95 °C for 10 min. Proteins (30 μg/lane) were loaded into the lanes of an SDS-polyacrylamide gel and separated by electrophoresis. The proteins were then transferred to PVDF membranes at 300 mA for 53 min. After blocking with 5% nonfat milk for at least 2 h, we incubated the membranes with anti-GSK-3β, pSer9-GSK-3β, H60, Rae1, pSer535-eIF2B, Mult-1, and β-actin antibodies overnight at 4 °C. After washing three times with 1 × TBST, the membranes were incubated with secondary antibody for 1 h. Density analysis was performed using Image J2, and the experiments were repeated three times.
Flow cytometry
ROS level was examined with a ROS assay kit that sets DCFH-DA as the probe. Then we detected the intensity of the DCF signal by flow cytometry to quantify the intracellular production of ROS. 4T1 cells were trypsinized, incubated with DCFH-DA-containing serum-free medium for 20 min at 37 °C, centrifuged at 1,000 rpm for 5 min, discarded the supernatant, and then, washed three times with PBS. Cells were analyzed on BD FACS Canto II. Data were analyzed with FlowJo 7.6.
Cytotoxicity assay
4T1 cells treated with TWS119, LY294002, H2O2, and NAC were incubated with Calcein-AM. Then, the cells were incubated with freshly isolated NK cells for 4 h at various effector/target ratios. The fluorescence of each supernatant was measured at an excitation peak of 490 nm and an emission peak of 515 nm using the multispectral spectrophotometer. The percentage of specific lysis was calculated according to the following formula: the rate of specific lysis (%) = [(experimental release-spontaneous release)/(maximum release-spontaneous release)] × 100%.
Transwell migration assay
The assay was performed in a 24-well Boyden chamber with 8-μm pore size (Corning, Shanghai, China), and 4T1 cells (1 × 105) were seeded in the upper chamber with TWS119 for 24 h and LY294002 for 16 h, we fixed the undersurface cells of transwell chambers with methyl alcohol and stained them with 0.1% crystal violet. A Nikon Eclipse 80i microscope and Nikon Elements Imaging System Software were used to capture images of migrated 4T1 cells.
Immunohistochemistry (IHC)
Formalin-fixed and paraffin-embedded samples were cut into 5-μm sections. Antigen retrieval was carried out by boiling in citrate buffer (pH 6.0), and the tissues were incubated with anti- GSK-3β, anti-pSer9-GSK-3β, anti-H60, anti-Rae1, and anti-pSer535-eIF2B antibody overnight, diaminobenzidine (DAB) was used for color development (Nikon Eclipse 80i). Visible nodules with weak or strong staining were counted under microscope. Mean densities in the plotted areas were quantitatively analyzed by the Image Pro Plus (IPP) software. More than ten views were used for the analysis each image, and three images of different treatment groups were selected randomly to perform analysis.
Mitochondrial ROS (mROS) measurement
mROS was measured using a MitoSOXTM Red mitochondrial superoxide indicator (YEASEN, Shanghai, China) according to the manufacturer's instructions. 4T1 cells were added in 24-well plate on the cover slip, after being treated with TWS119, H2O2, and NAC for 24 h and LY294002 for 12 h. Cells of all groups were incubated with 5 μM MitoSOXTM reagent working solution for 10 min at 37 °C in the dark. After washing three times with warm HBSS, we detected the ROS intensity with a fluorescent microscope.
Quantifying the activity of MRCC I/III
MRCC I/III kits (Solarbio, Beijing, China) were used according to manufacturer’s instructions. 5 × 106 4T1 cells were collected to homogenate on ice, and the homogenate was then centrifuged at 600 rpm, 4 °C for 5 min, then we suspended the precipitant (mitochondria), and treated them by ultrasonication in ice bath (200 W, 3 s for 30 times, interval for 10 s). Forty microliters of samples were mixed with 800 μL working solution, and recorded the initial absorbance (A1) and final absorbance after 2 minutes (A2) at 340 nm and 550 nm for MRCC I/III, respectively. The activity unit is defined as 1 nmol NADH or reduced CYC consumed by 104 cells per minute, and the activity was calculated as per the formulae: (1) activity of complex I (U/104 cells) = 0.73 (A1–A2), and (2) activity of complex III (nmol/min/104 cells) = 0.248 (A1–A2).
Statistical analysis
The data are presented as “mean ± SE” and analyzed using SPSS 13.0. The differences between two groups were assessed by Student's t-test and the differences among three or more groups by one-way ANOVA followed by the Dunnett test. P < 0.05 is considered statistically significant.
Results
GSK-3β inactivation promoted breast cancer growth and lung metastasis by suppressing the function of NK cells
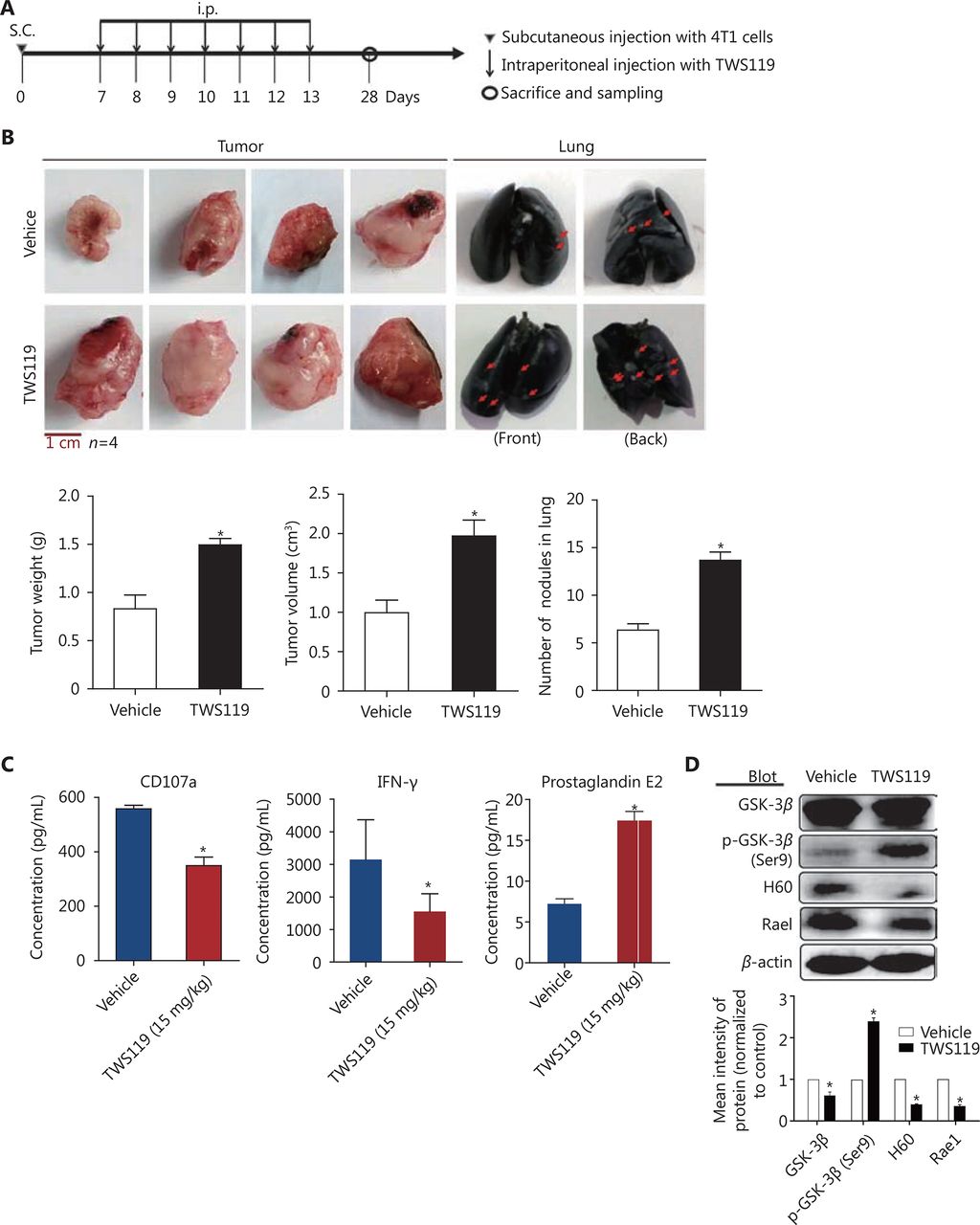
To investigate the effects of GSK-3β on breast cancer and cytotoxicity of NK cells, we performed a series of experiments in 4T1 cell-harboring mice treated with TWS119 (4,6-disubstituted pyrrolo-pyrimidine), which functions by phosphorylating GSK-3β at Ser9. After the formation of subcutaneous transplanted tumors, BABL/C mice were intraperitoneally injected with TWS119 (15 mg/kg) once a day, continuously for seven days, while the control group mice were injected with normal saline at the same time points. The mice were sacrificed at the 28th day after the initial inoculation of 4T1 cells (Figure 1A). The tumors of the treatment group were significantly larger than those of the control group, and the number of lung metastatic nodules, both in the front and back of the lung, also increased significantly compared to that in the control group (*P < 0.05, Figure 1B). Then, to evaluate the cytotoxicity indirectly, we employed ELISA assay and the results showed that the levels of NK cell cytotoxic factors, CD107a and IFN-γ, decreased significantly in the TWS119 treatment group, and in contrast, the levels of inhibitory molecule PGE2 in NK cells increased (*P < 0.05, Figure 1C). NK cells target the NKG2DLs of the tumor cells, and eliminate them. Therefore, we detected the expression of NKG2DLs by Western blot in fresh tumor tissue homogenate, and similarly, found that the levels of H60 and Rae1 in the TWS119 treatment group were attenuated compared with that in the control group (Figure 1D). Taken together, these data indicated that inactivated GSK-3β could promote breast cancer growth and lung metastasis partly by suppressing the function of NK cells and the response of tumor cells to NK cells.
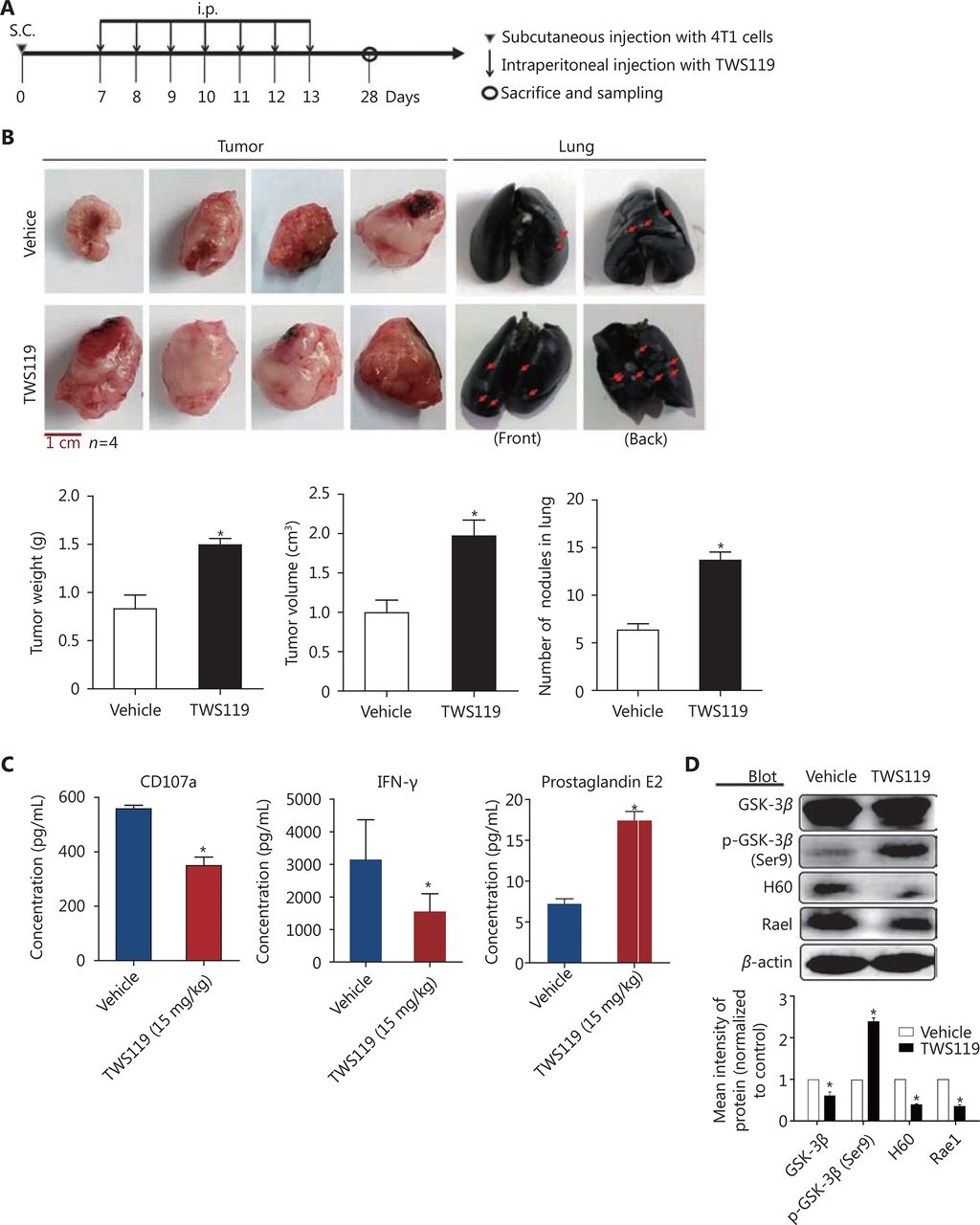
Inhibition of GSK-3β promoted the tumor growth and suppressed NK function in 4T1 transplanted tumor mice. (A) Tumor transplantation method to establish mouse breast cancer animal model. (B) Representative images and quantification of tumor weight, tumor volume, and lung metastasis nodules in TWS119 (15 mg/kg)-treated tumor-bearing mice. (C) ELISA analysis of CD107a, IFN-γ, and PGE2 levels in eyeball blood serum. *, indicates values with P < 0.05 vs. vehicle. (D) The expression of NKG2D ligands (H60 and Rae1) was analyzed by Western blot using tumor tissue homogenate.
pSer9-GSK-3β downregulated the expression of NKG2DLs and promoted the migration of 4T1 cells in vitro
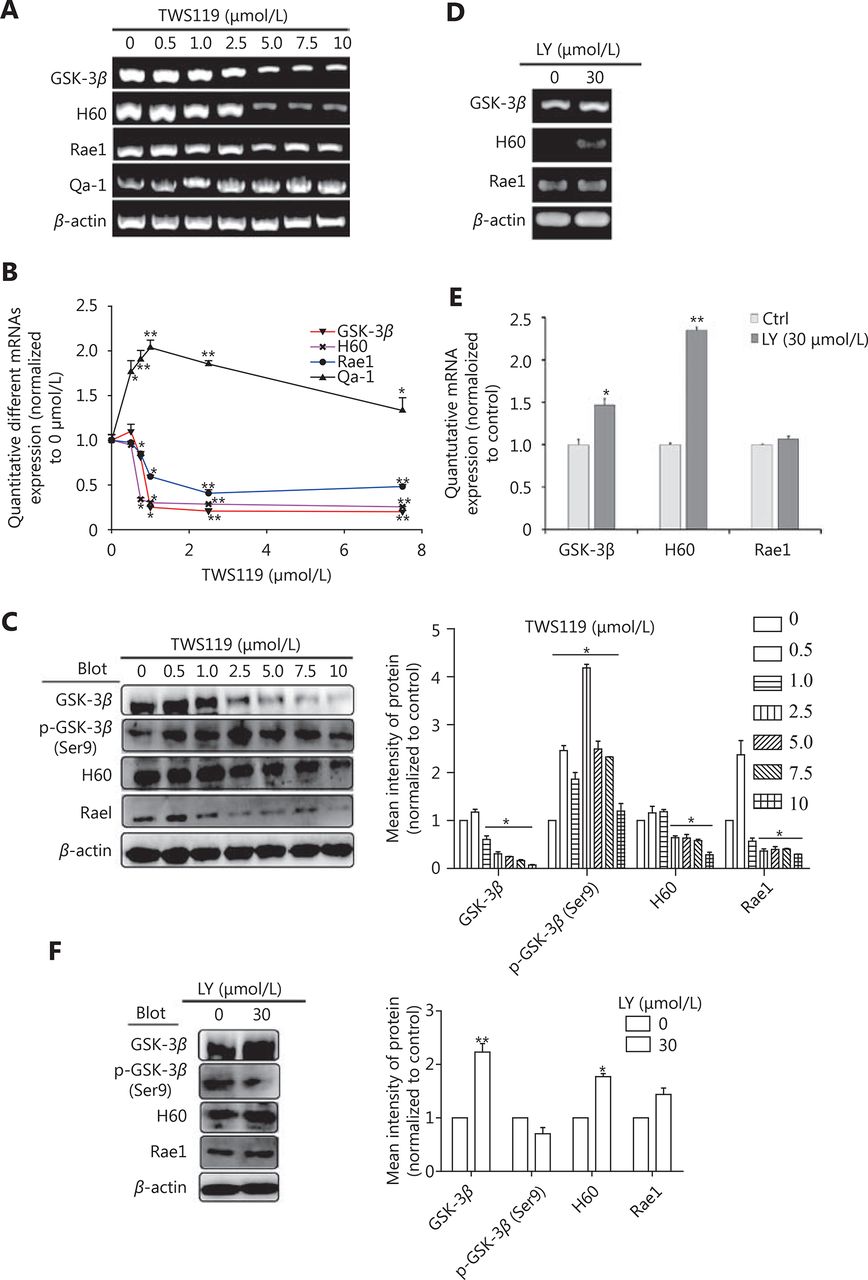
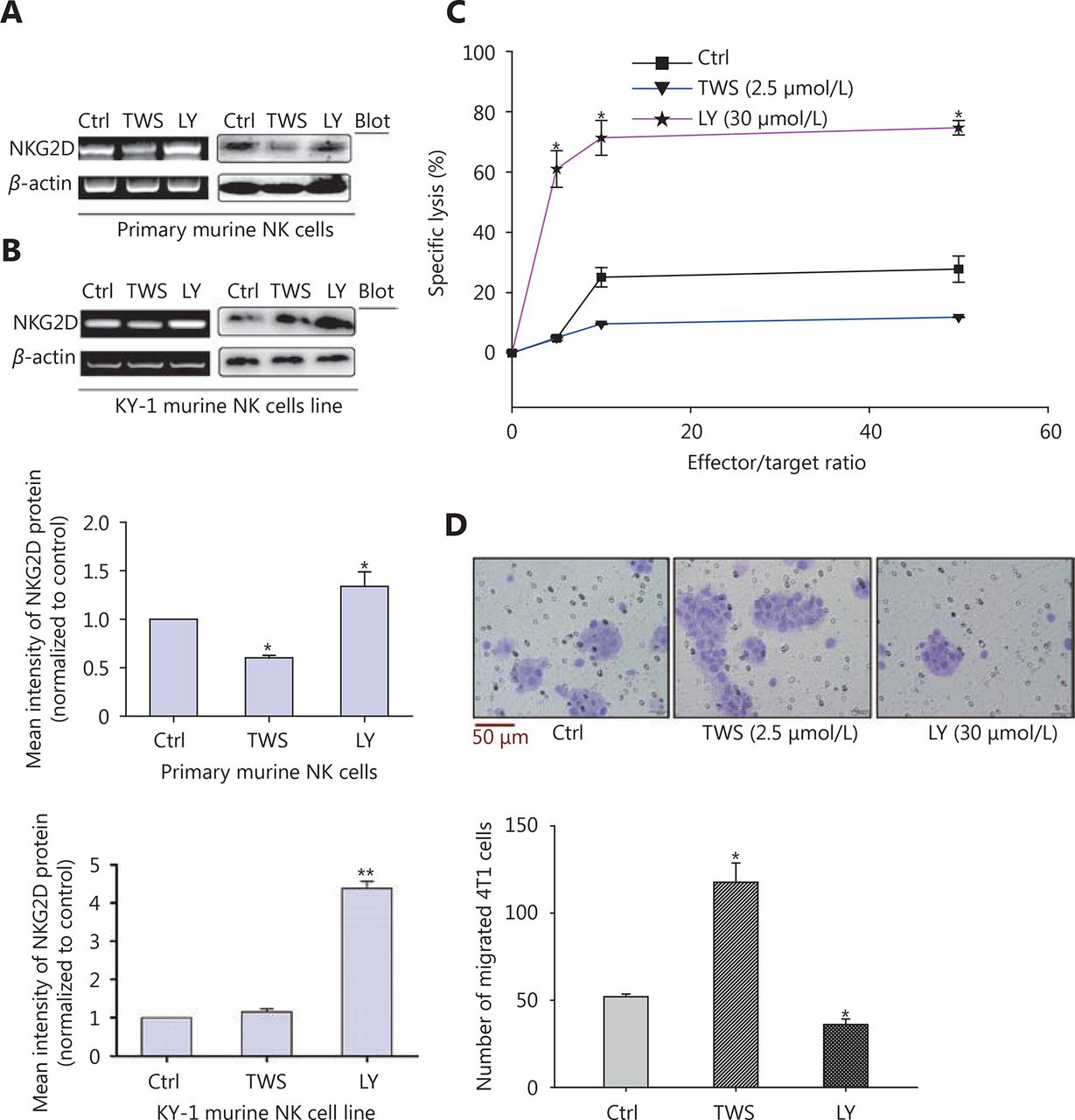
TWS119 suppressed the function of NK cells, and promoted the growth and metastasis of breast cancer by phosphorylating GSK-3β at Ser9. To further explore the role of GSK-3β in breast cancer, we cultured 4T1 cells with different concentrations of TWS119. Our results revealed that the expression of NKG2DLs (H60 and Rae1) was downregulated after TWS119 treatment in concentration-dependent manner at both mRNA and protein levels; 2.5 μmol/L TWS119 was selected as the optimum concentration based on the degree of GSK-3β phosphorylation as detected by RT-PCR (Figure 2A), real time PCR (Figure 2B) and Western blot analysis (Figure 2C, *P < 0.05, ** P < 0.01). LY294002, a PI3K/Akt pathway inhibitor, exhibits the ability to decline the phosphorylation level as mentioned above, and was speculated to be an indirect activator of GSK-3β. We found that 4T1 cells treated with 30 μmol/L LY294002 significantly upregulated the expression of the NKG2DLs at both on mRNA and protein levels as observed with RT-PCR ( Figure 2D), Real time PCR (Figure 2E), and Western blot analysis (Figure 2F, *P < 0.05, ** P < 0.01). The supernatant obtained from TWS119- and LY294002-treated 4T1 cells was used to culture primary NK cells ( Figure 3A)and KY-1 cell line (Figure 3B), and results of RT-PCR and Western blot analysis showed that the expression of NKG2D was downregulated after inhibiting the GSK-3β and was upregulated after the GSK-3β was indirectly activated. In addition, we performed a Calcein AM release assay to determine the effects of GSK-3β on the cytotoxicity of NK cells. Briefly, primary NK cells were obtained from spleen of BALB/C mice by magnetic-activated cell-sorting, and then co-incubated with 4T1 cells at different effector/target ratios (E/T ratio). We observed that TWS119 lowered the activity of NK cells, while LY294002 significantly promoted their activity (*P < 0.05, Figure 3C). Furthermore, transwell migration assay validated that pSer9-GSK-3β contributed to the migration of 4T1 cells in vitro (*P < 0.05, Figure 3D), which was consistent with the results of the lung metastases in animal experiments. Therefore, we concluded that the inhibition of PI3K/Akt/GSK-3β signaling promoted the cytotoxicity of NK cells and upregulated the expression of NKG2DLs on the surface of 4T1 cells, thereby suppressing the aggressiveness of tumor cells.
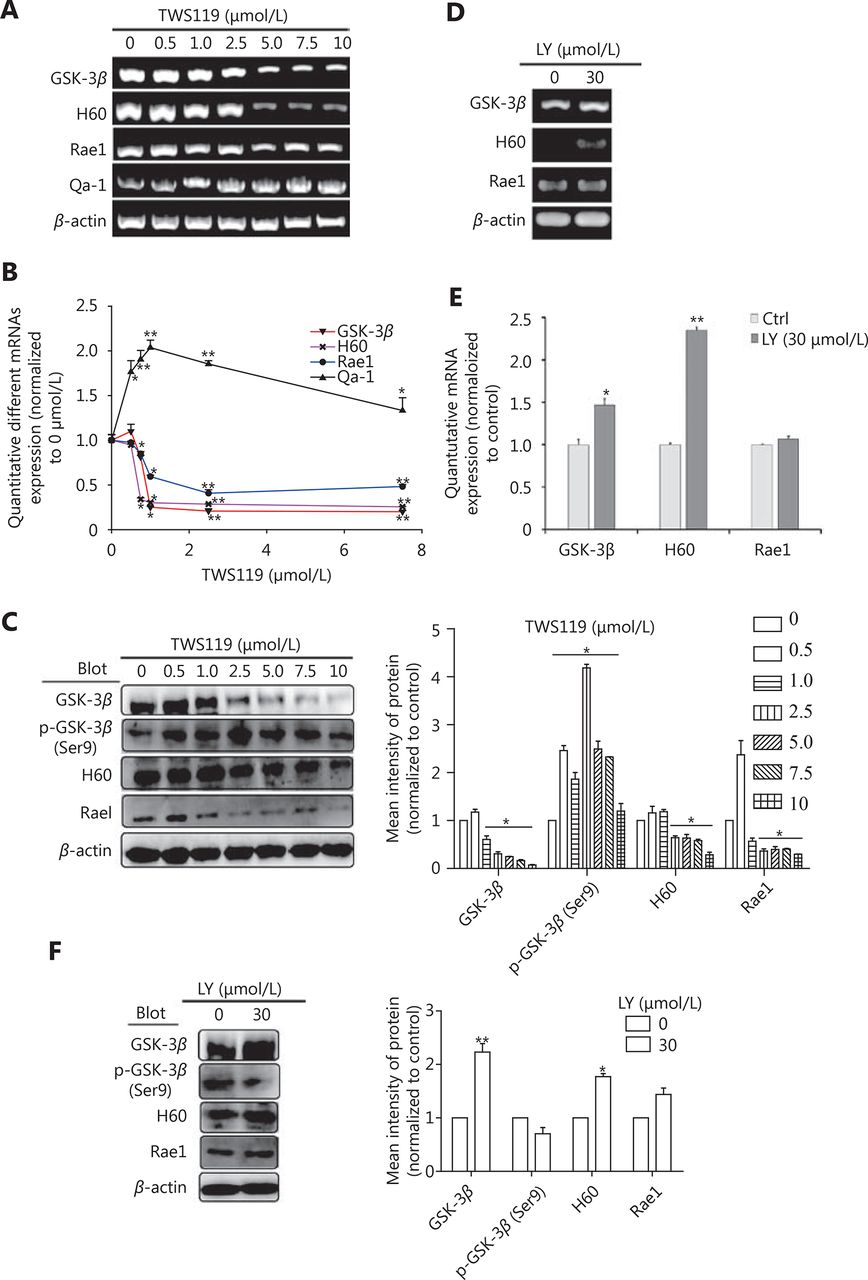
GSK-3β upregulated the expression of NKG2D ligands. PCR (A) and Western blot (C) were performed in 4T1 cells to determine the expression of H60 and Rae1 or Qa-1 after treatment with inhibitor TWS119 (2.5 μmol/L). (B) Quantitative analysis of the PCR in 2A. PCR (D) and Western blot (F) were performed in 4T1 to determine the expression of H60 and Rae1 after treatment with GSK-3β activator, LY294002 (30 μmol/L). (E) Quantitative analysis of the PCR in 2D. *, ** indicate values of P < 0.05, P < 0.01, respectively.
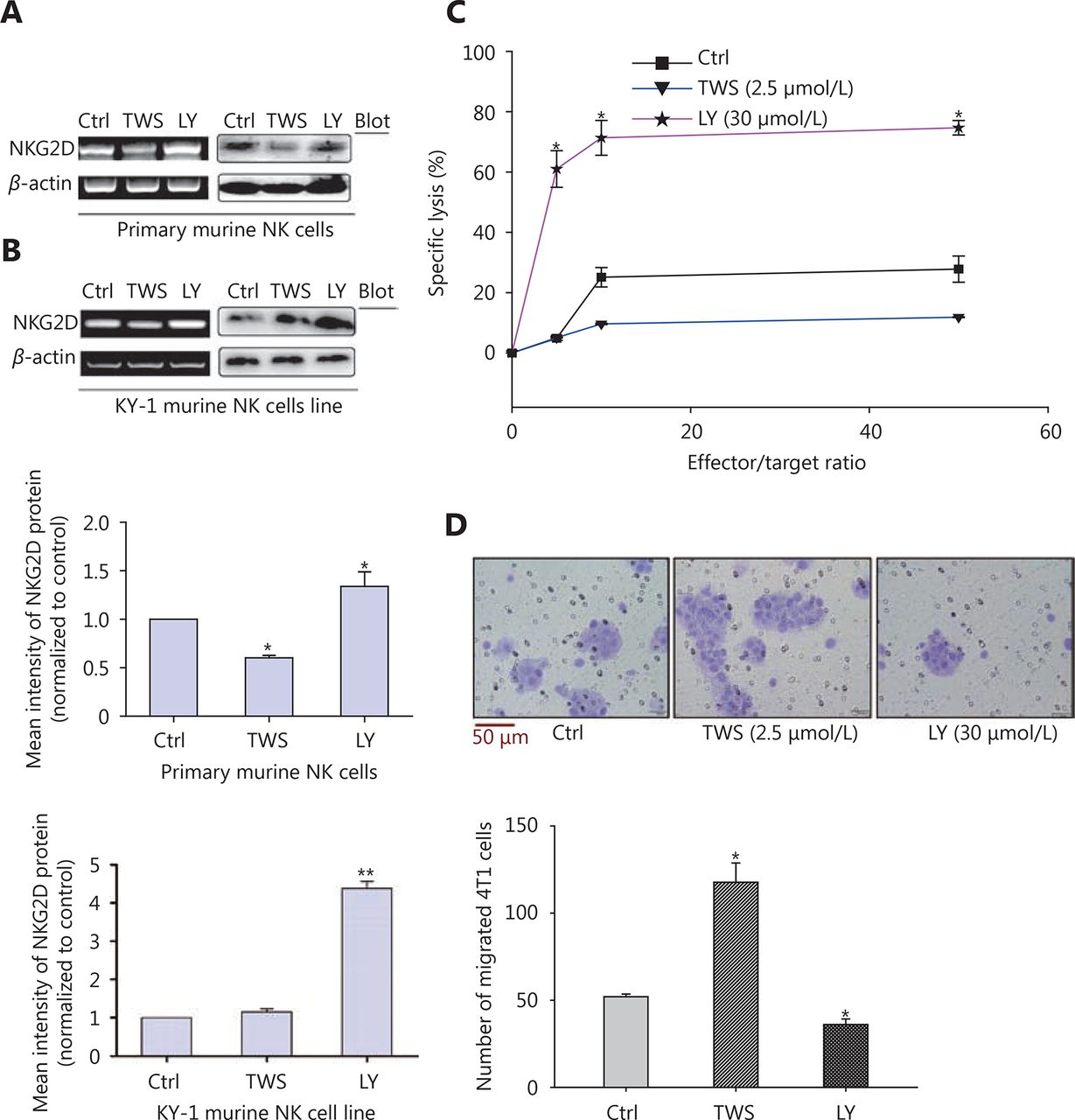
GSK-3β improved NK cell cytotoxicity and suppressed migration of 4T1 cells by upregulating the expression of NKG2D. After the treatment with TWS119 and LY294002, the expression of NKG2D in primary NK cells (A) and in mice NK cell line KY-1 (B) was assessed by RT-PCR and Western blot. (C) The cytotoxicity of NK cells was measured by Calcein AM-release assay. (D) The migration ability of 4T1 cells was detected by transwell migration assay. The migrated cells were quantified for 4 random fields. *, ** indicate values of P < 0.05, P < 0.01, respectively.
Intracellular ROS mediated the GSK-3β induced regulation of NK and 4T1 cells
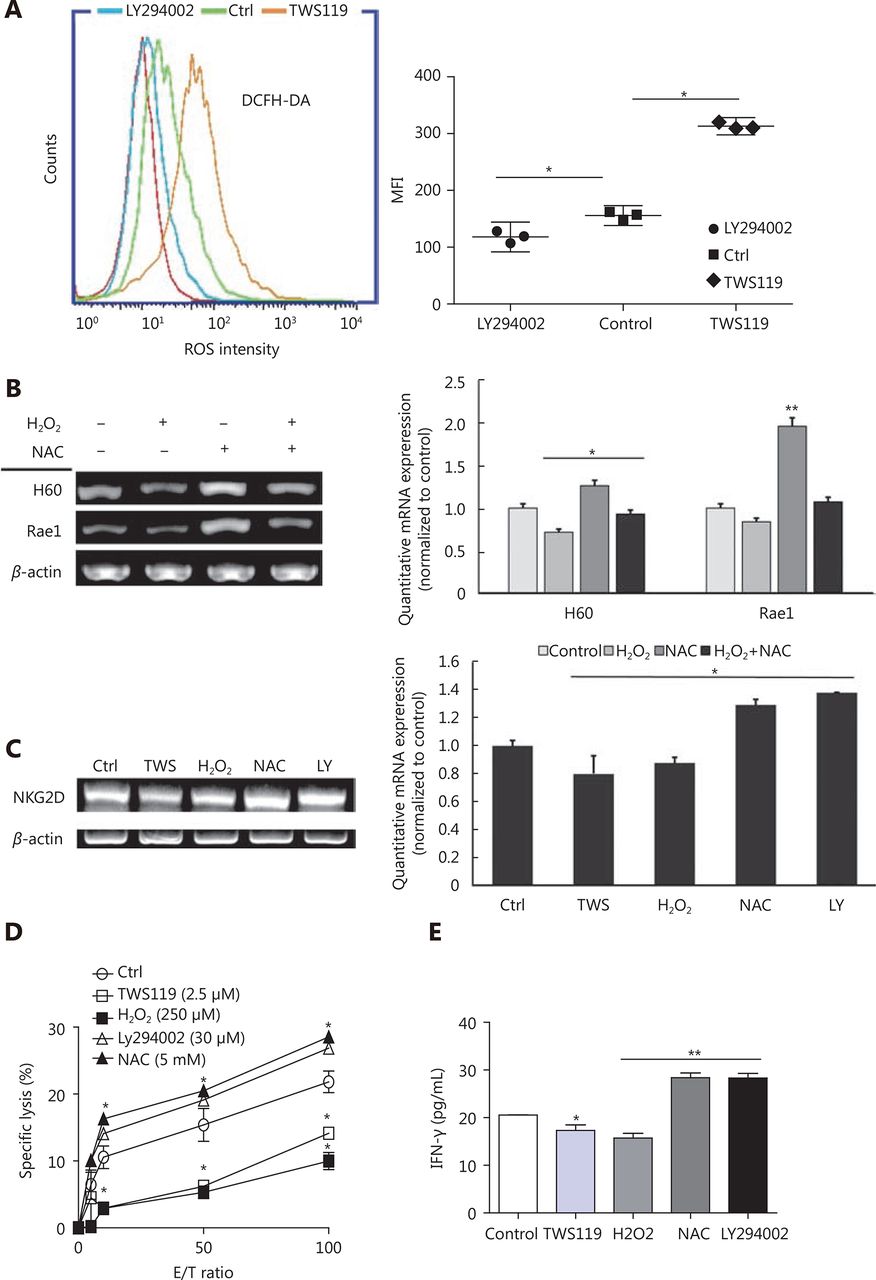
Accumulating evidence has reported that GSK-3β function in cancer via various classical signaling pathways, including Wnt/β-catenin, Ras/Raf/MEK/ERK, Notch, Hedgehog, and so on19,43. It has also been shown that chronic ROS enhanced the growth and tumorigenic potential of the MCF7 breast cancer and it even activated various signaling pathways, such as PI3K/AKT38. Therefore, we tried to further explore the correlation between ROS and GSK-3β. The data obtained from the flow cytometry analysis showed that the intracellular ROS change tendency was significantly different between TWS119 and LY294002-treated groups (*P < 0.05, Figure 4A). Compared with that in the control group, TWS119 inactivated GSK-3β and led to a higher ROS level.
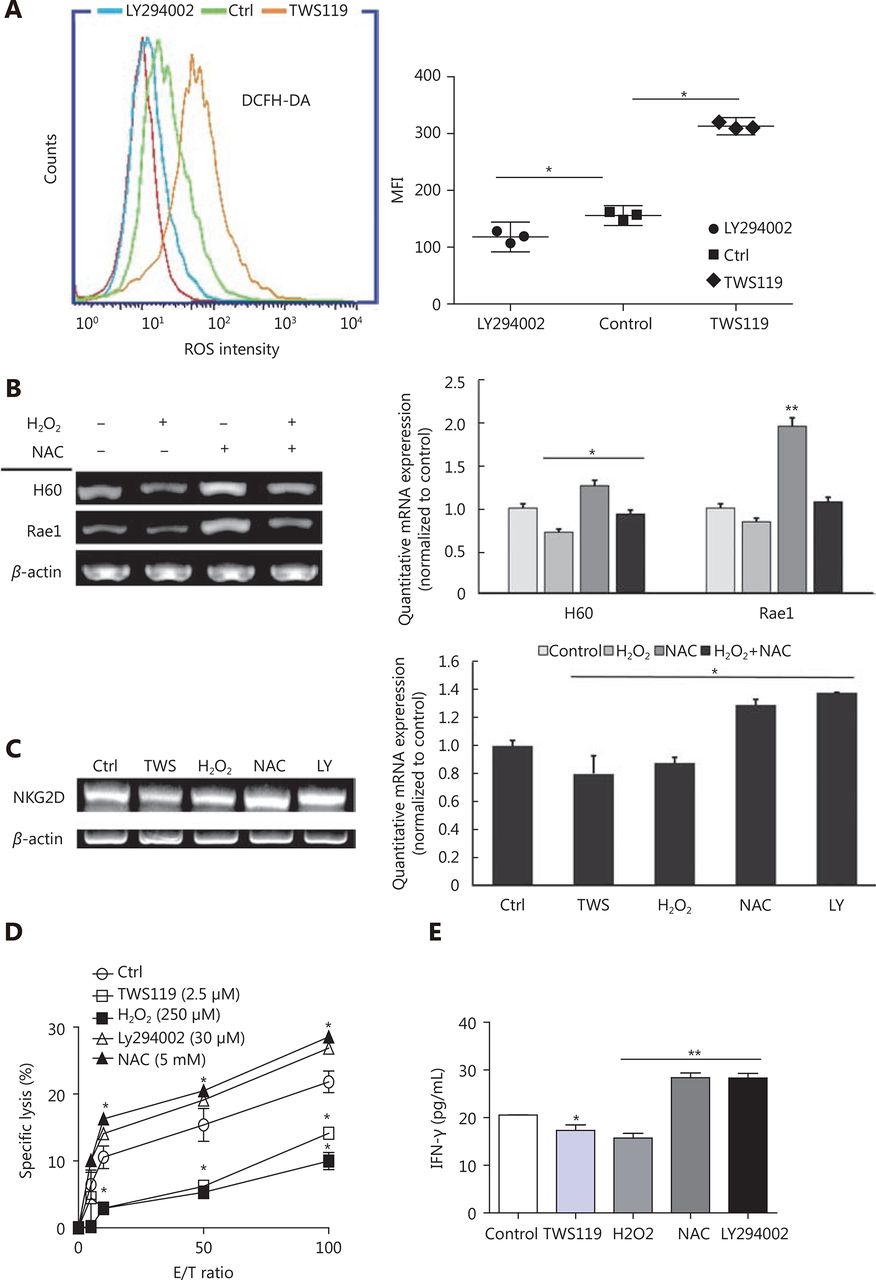
GSK-3β enhanced NK cell function by attenuating the intracellular ROS production. (A) ROS levels of 4T1 cells, treated with TWS119 and LY294002, were determined by flow cytometry. (B) The expression of H60 and Rae1 was analyzed by RT-PCR and real time-PCR after H2O2 (250 μmol/L) and NAC (5 mmol/L) treatment. (C, D, E) The expression of NKG2D, the cytotoxicity of NK cells, and IFN-γ concentration was evaluated by RT-PCR, real time-PCR, Calcein release assay, and ELISA. *, ** indicate values of P < 0.05, P < 0.01, respectively.
Does ROS affect the activity and the sensitivity of NK cells, at least partly? H2O2 (250 μmol/L) and N-acetylcysteine (NAC, a thiol antioxidant, 5 mmol/L) were used to induce and eliminate ROS, respectively, to answer this question. By RT-PCR and real time-PCR, we first detected the NK cell activation related ligands (H60 and Rae1) in different groups, and found that NAC could upregulate the expression of H60 and Rae1, which was contrary to the observation in the TWS119 group (*P < 0.05, ** P < 0.01, Figure 4B). Then, the expression of NKG2D in primary NK cells was downregulated in TWS119- and H2O2-treated groups, and was upregulated in LY294002- and NAC-treated groups (*P < 0.05, Figure 4C). Combined with the results from Figure 3A, we speculated ROS to be an important factor regulated by GSK-3β, and ROS influenced the recognition and combination of NK cells to 4T1 cells via NKG2D-NKG2DLs. We also discovered that pSer9-GSK-3β damaged the activity of NK cell via ROS. The results of Calcein release assay showed that TWS119 and H2O2 attenuated the direct cytotoxicity of NK cells while LY294002 and NAC enhanced their cytotoxicity (Figure 4D). Further, IFN-γ release, an indirect index of the NK cell cytotoxicity, exhibited the same trend (*P < 0.05, ** P < 0.01, Figure 4E). To conclude, ROS was responsible for GSK-3-induced regulation of NK and 4T1 cells; therefore, activating GSK-3β or inhibiting ROS could prove to be a potential anti-breast cancer therapy.
Mitochondrial ROS (mtROS) was mainly responsible for the regulation of NK cells by GSK-3β
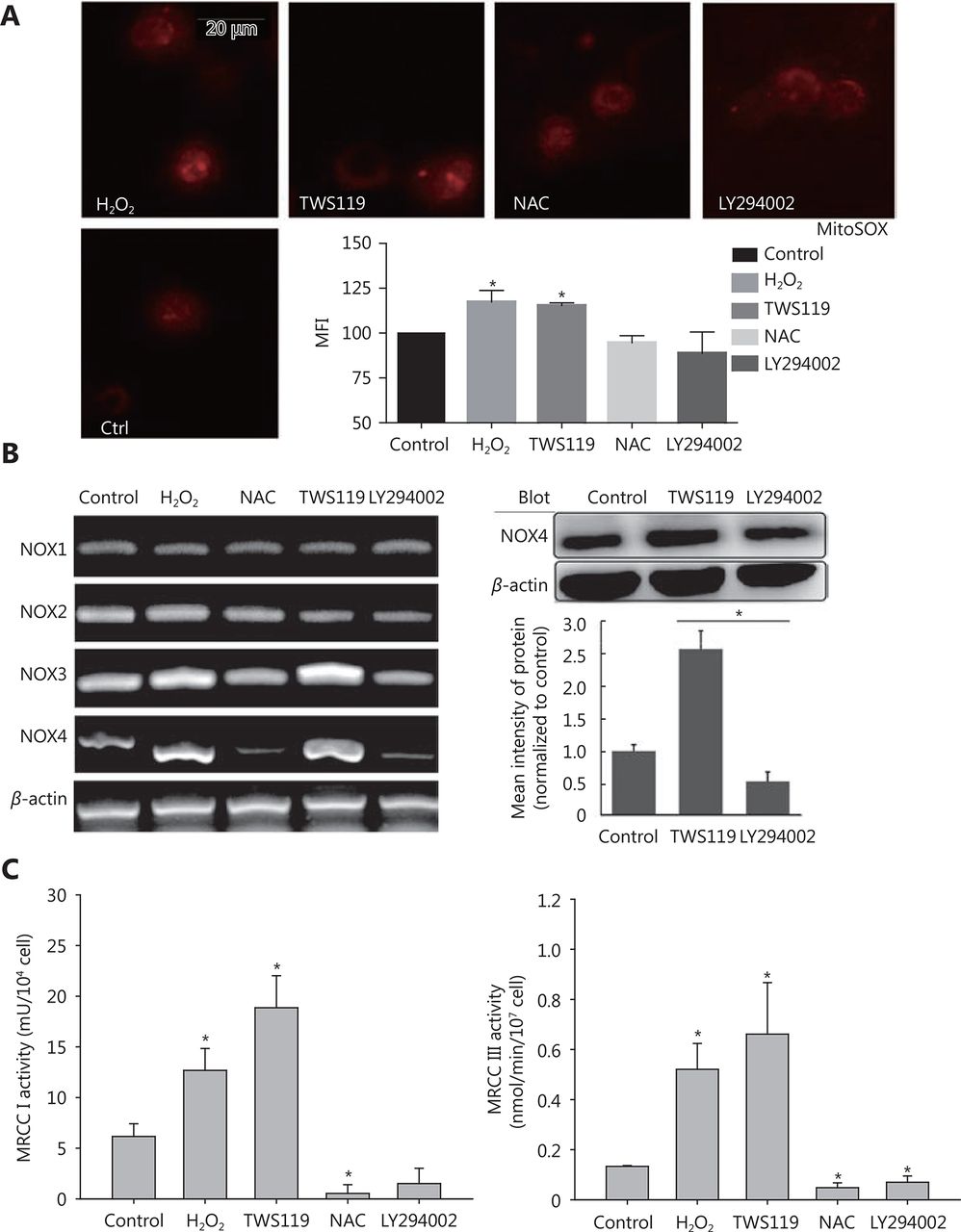
Commonly, mitochondria are the main sources of ROS. Therefore, we measured the potential changes in mitochondrial ROS. Firstly, we observed the distribution of mitochondrial ROS with a fluorescent dye, MitoSOX red, which specifically targets mitochondrial ROS. The results showed that TWS119 and H2O2-treated groups significantly enhanced mtROS production, while NAC and LY294002 attenuated it. This change in mtROS level was in agreement with the change in intracellular ROS level, and the mtROS displayed a strong co-localization with mitochondria. Our results showed that ROS was indeed responsible for the dysfunction of mitochondria (*P < 0.05, Figure 5A).
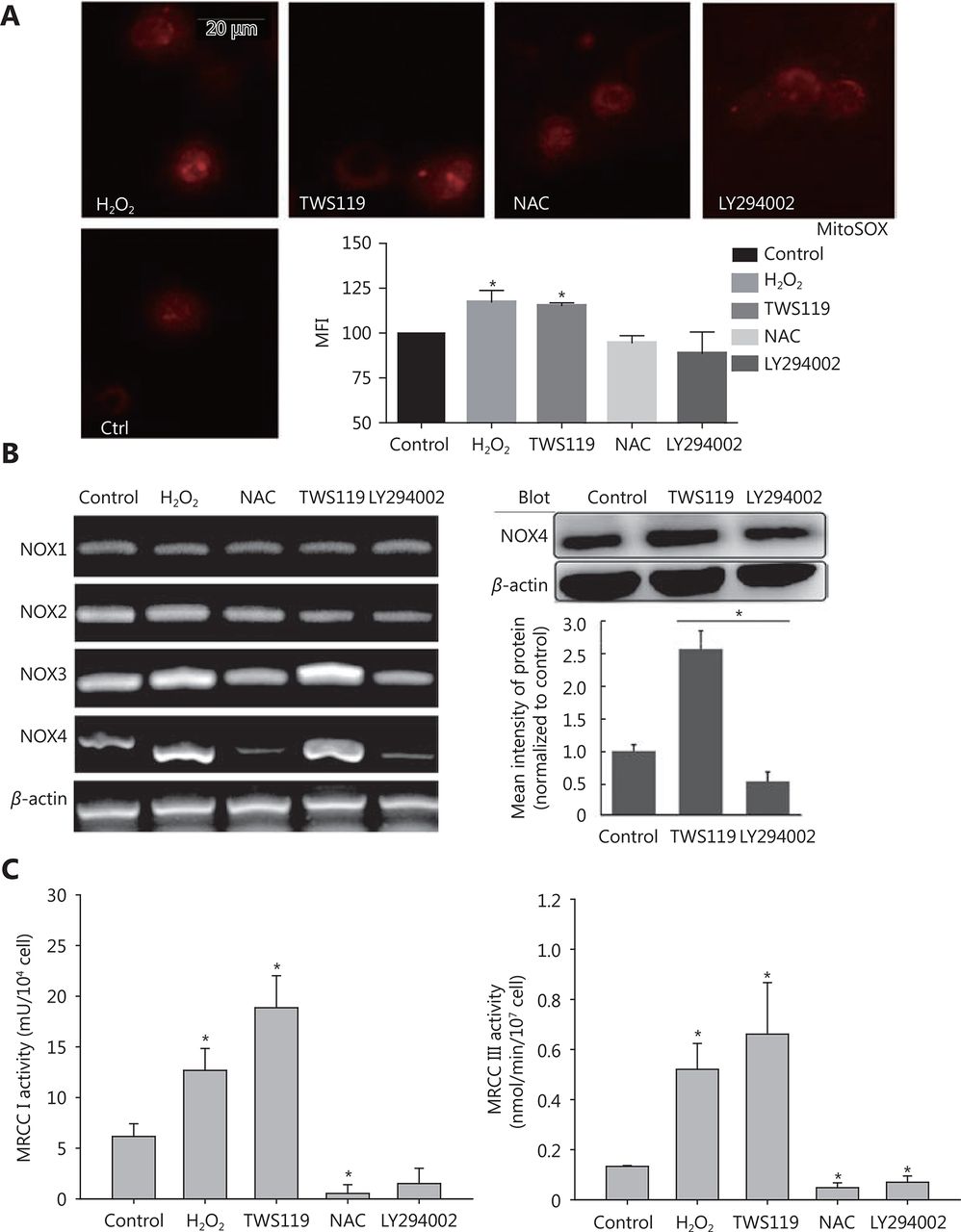
Attenuating mitochondrial ROS was mainly responsible for the improved function of NK cells after GSK-3β treatment. (A) MitosoxTM fluorescence probe was used to observe mitochondrial ROS activity after treatment with TWS119, LY294002, H2O2, and NAC. (B) The expression of NOX1-4 was amplified by RT-PCR and NOX4 levels were further analyzed by Western blot. (C) The activity of MRCC I and III in 4T1 cells was determined by a multiscan spectrum microplate spectrophotometer. *, ** indicate values of P < 0.05 and P < 0.01, respectively.
Mitochondria produce ROS mainly via NOX and mitochondrial respiratory chain complex. In this study, using RT-PCR amplification and Western blot analysis, we initially found that NOX3 and NOX4 upregulated the expression by 1.5-fold and 2.3-fold on mRNA level, respectively. We also found NOX4 protein was upregulated after TWS119 and downregulated after LY294002 treatment (*P < 0.05, Figure 5B), while NOX1 and NOX2 induced no such variation (data not shown). Besides, the activity of mitochondrial respiratory chain complex (MRCC) represents the electron flux during redox reaction, and partially reflects mitochondrial ROS status to some degree. By detecting the activity of MRCC, we indirectly evaluated the effect of GSK-3β on ROS level, and this investigation proved that pSer9-GSK-3β and H2O2 significantly upregulated the activity of MRCC I and III, whereas LY294002 and NAC downregulated the activity of MRCC I and III (*P < 0.05, Figure 5C).
pSer535-eIF2B was involved in the regulation of NK cells and tumor cells followed by GSK-3β/ ROS
GSK-3β is a multifunctional molecule that regulates a variety of signaling pathways. Previous studies have revealed that CCND1 (cyclinD1), NFAT1 (nuclear factor of activated T cells), and eIF2B exhibited high expression levels in breast cancer cells and tissues, and the activated GSK-3β phosphorylated these downstream molecules (CCND1, NFAT1, and eIF2B)44-46to suppress the growth and metastasis of breast cancer.
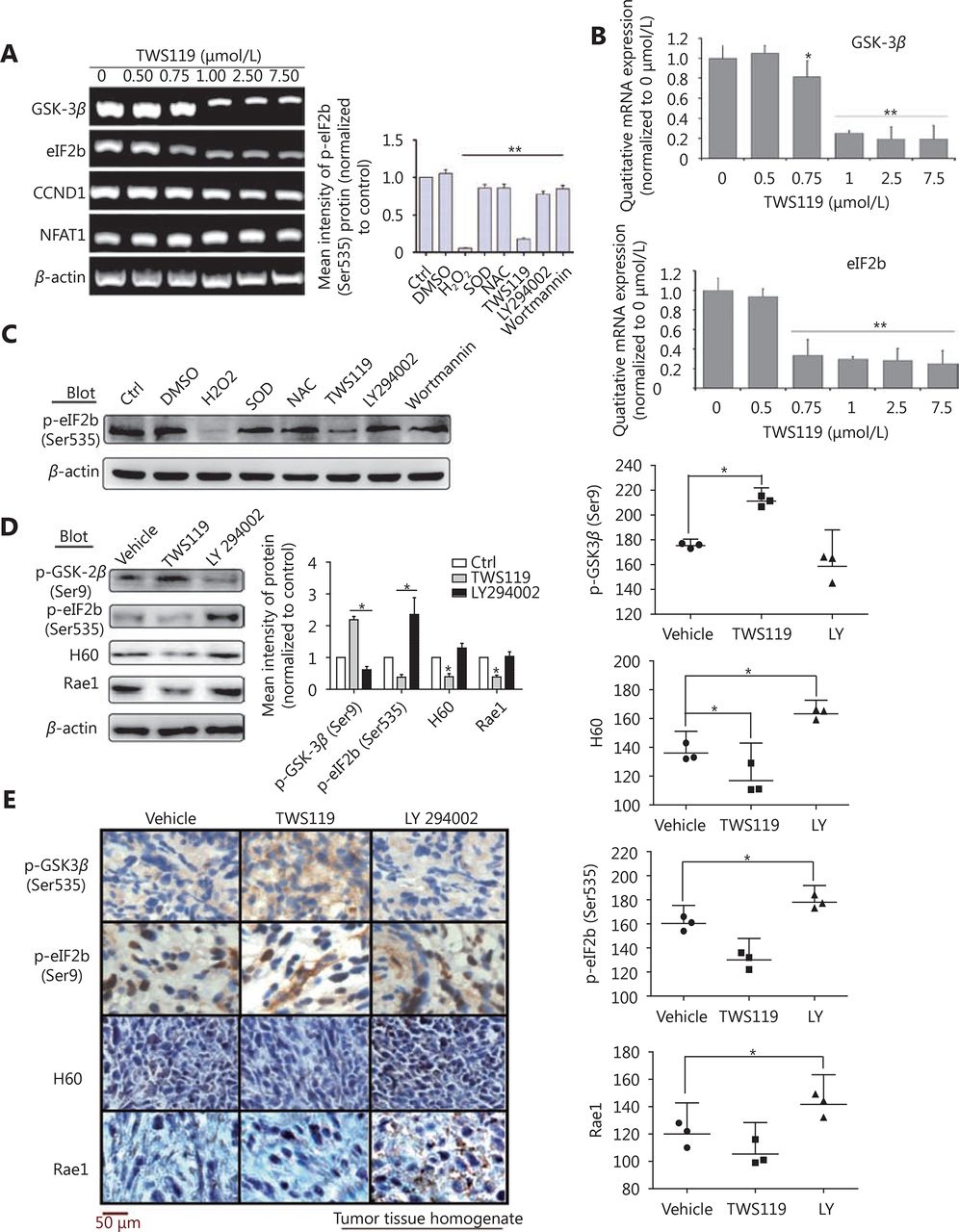
Furthermore, we treated 4T1 cells with different concentrations of TWS119, performed RT-PCR and real time-PCR, and the results showed that eIF2B gradually decreased after the TWS119 treatment (*P < 0.05, ** P < 0.01, Figure 6A and 6B); however, the change in CCND1 and NFAT1 was obscure (data not shown). Study showed that ROS could inhibit the phosphorylation of eIF2B by oxidative modification of serine residue of ε subunit47, which may partly explain how GSK-3β affects eIF2B. We next found the expression of pSer535-eIF2B significantly decreased in TWS119-treated 4T1 cells, which was consistent with the tendency of the H2O2 group, whereas LY294002-, wortmannin-, NAC-, and SOD-treated groups exhibited opposite effects on the expression of pSer535-eIF2B (Figure 6C). These results further suggested that pSer535-eIF2B might be the downstream regulatory factor of GSK-3β/ROS. Does pSer535-eIF2B affect the expression of NKG2DLs? We analyzed the tumor tissues obtained from animal experiments and found that, with the phosphorylation level of GSK-3β decreased, the phosphorylation of eIF2B at Ser535 and the expression of H60 and Rae1 increased by Western blot analysis (Figure 6D) and IHC test (*P < 0.05, Figure 6E).
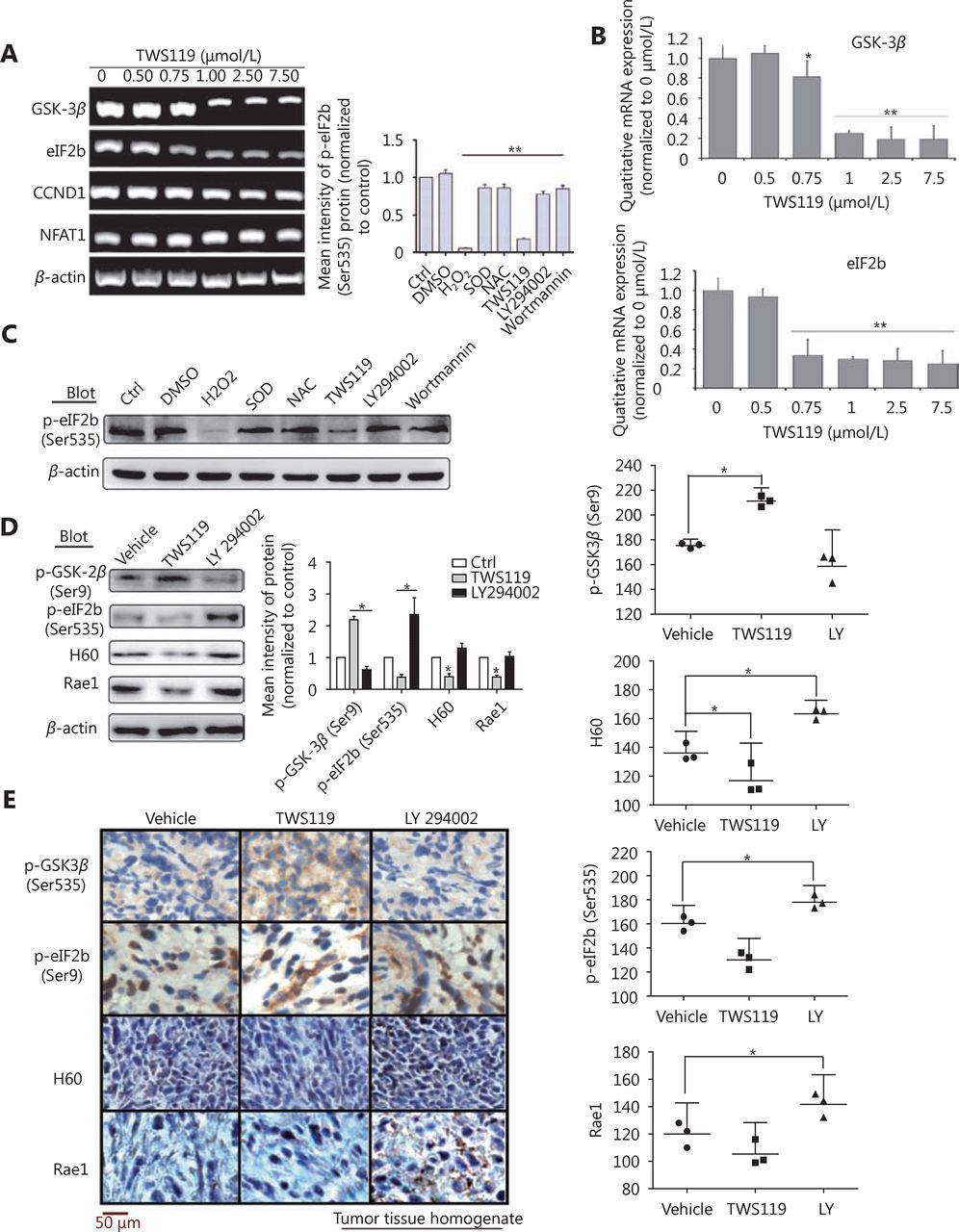
eIF2B functions as a downstream molecule of PI3K/AKT/GSK-3β/ROS pathway to upregulate NKG2D ligands. (A) eIF2B, CCND1, and NFAT1 levels were investigated by RT-PCR. (B) mRNA levels of GSK-3β and eIF2B, protein levels of pSer9-GSK-3β, pSer535-eIF2B, H60, and Rae1 were quantified. (C) The expression of p-eIF2B induced by H2O2, NAC, and SOD (1,000 U/mL) as well as TWS119, LY294002, and Wortmannin (2 μmol/L, another inhibitor of PI3K/AKT) was detected by Western blot. (D) The protein levels of p-GSK-3β, p-eIF2B, H60 and Rae1 expressed in tumor tissue homogenate after intraperitoneal injection of TWS119 and LY294002 (5 mg/kg) was assayed by Western blot. (E) Consecutive sections of above tissues were stained by DAB. The mean densities in the positive areas were quantitatively by the IPP software. *, indicates values of P < 0.05.
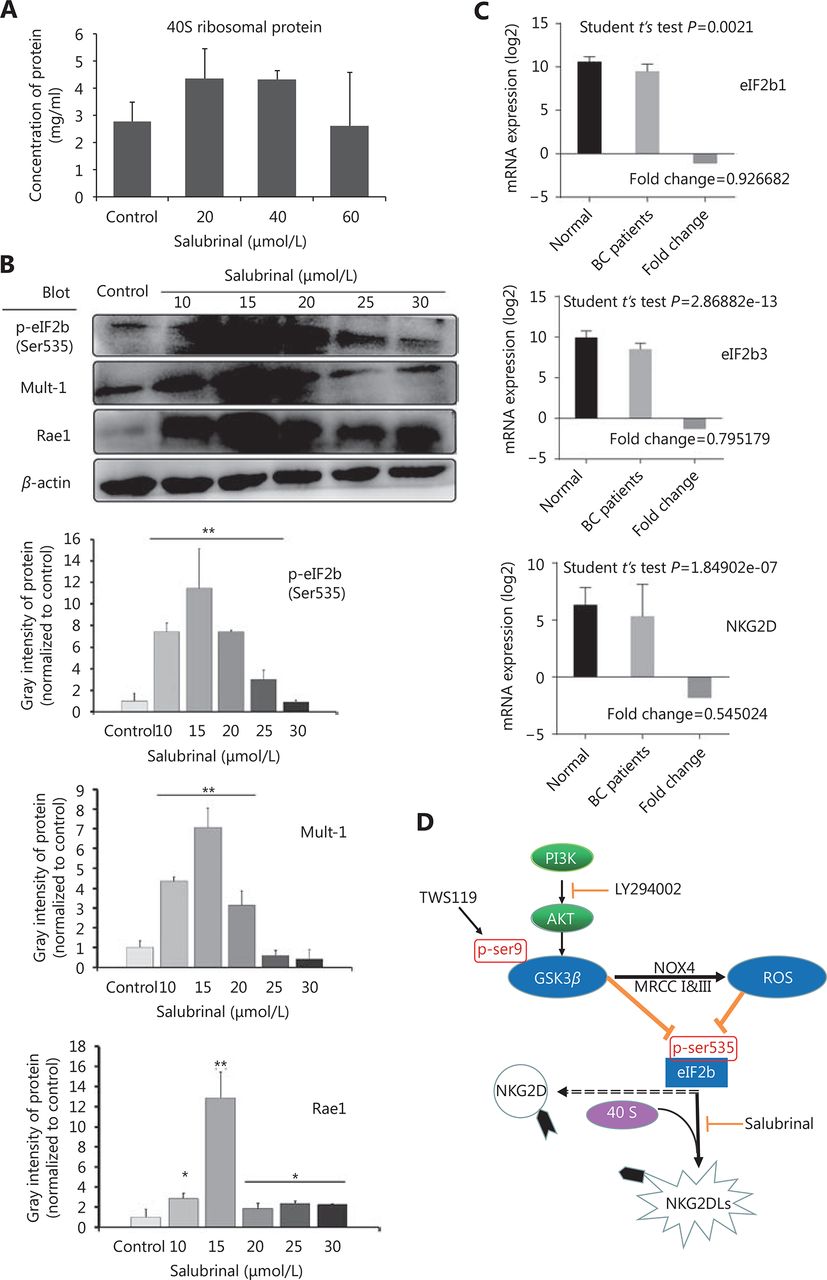
Therefore, we speculated that eIF2B might be one of the factors through which GSK-3β regulated the expression of NK cell ligands. To better understand the effect of eIF2B on NKG2DLs (Mult-1 and Rae1), we employed salubrinal, an eIF2α dephosphorylation inhibitor, to combine and competitively inhibit the activity of eIF2B. Normally, eIF2 interacts with initiator Met-tRNA, forms the trimeric complex after recruitment by the RPS19 protein of the ribosomal 40S subunit, and binds to ribosomal 60S subunit. After treatment with salubrinal, Ser51 of eIF2α subunit is phosphorylated, eIF2B becomes inactivated, the ribosomal 60S subunit stops synthetizing, and finally, leads to the 40S reservoir.
To confirm the efficacy of salubrinal, we examined the levels of ribosomal protein 40S by mouse RPS19 ELISA, and found the 40S retention was significantly enhanced after treatment with salubrinal (20 and 40 μmol/L) (*P < 0.05, Figure 7A). The results of Western blot showed that the expression of Mult-1 and Rae1 was upregulated with increase in the phosphorylation of eIF2B after treatment with salubrinal in a concentration-dependent manner and 15 μmol/L was optimal concentration (*P < 0.05, ** P < 0.01, Figure 7B). Given that eIF2B facilitates NK cells to kill tumor, in part because of robust expression of NKG2D and NKG2DLs, how does eIF2B affect the expression of NKG2D? Meta-analysis of 1023 breast cancer patients cataloged in StarBase v2.0 revealed a marked reduction in NKG2D mRNA levels with a decrease in expression of eIF2B1 and eIF2B3 (Figure 7C). We concluded that eIF2B influenced NKG2D despite the study number was relatively small (breast cancer patients: 998 samples vs. normal: 108 samples).
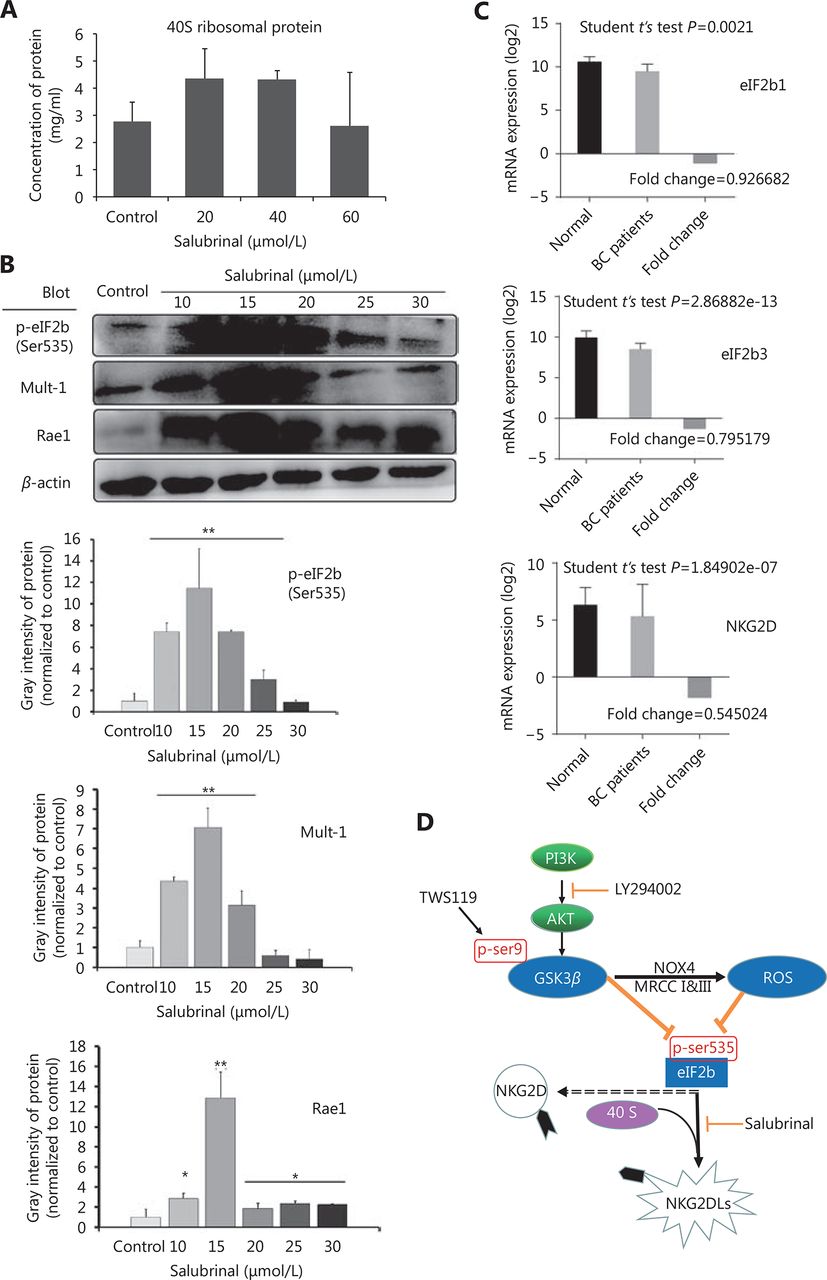
The cytotoxicity of NK cells was improved by inactivation of eIF2B. (A) The expression of RPS19 (a part of the ribosomal 40S subunit) was detected by ELISA to confirm the inhibition to eIF2B in the 4T1 cells after treatment with salubrinal. (B) The expression of NKG2D ligands (Rae1 and Mult-1) was upregulated after phosphorylation of eIF2B as detected by Western blot. (C) Meta-analysis showed that eIF2B1, eIF2B3, and NKG2D mRNAs expressed constitutivelyin human breast cancer patients (998 samples) and normal tissues (108 samples). (D) Schematic model of this study. *, ** indicate values of P < 0.05, P < 0.01, respectively.
Finally, we can summarize that PI3K/Akt phosphorylated and inactivated GSK-3β at Ser9, the inactivated GSK-3β increased mitochondrial ROS, and subsequently, attenuated phosphorylation of eIF2B at Ser535, the rate-limiting factor of translation initiation, which might suppress the activity of NK cells by downregulating the expression of NKG2D and NKG2Dls (H60, Rae1, or Mult-1), and finally, promoted the growth and lung metastasis of mouse breast cancer (Figure 7D).
Discussion
GSK-3β has been reported as both a tumor promoter as well as suppressor in different cancers, and is abnormally inactivated in breast cancer. In this study, the animal experiments demonstrated that inhibiting GSK-3β by TWS119 promoted the growth and metastasis of mouse breast cancer, which was consistent with results of a previous study20. The tumor progression is regulated by the immune cells in tumor microenvironment, and we found that inactivated GSK-3β attenuated the activity of NK cell. In vitro studies also found that GSK-3β regulated the cytotoxicity of NK cells and susceptibility of 4T1 cells to specific cytotoxicity.
Previous research has indicated that Akt activation led to the inactivation of GSK-3β, which was related with poor clinical outcome21. As upstream molecules, both Akt and PKC can inhibit GSK-3β by phosphorylating Ser9, and it has been reported that LY317615, a selective inhibitor of PKC, could effectively suppress the phosphorylation of GSK-3β at Ser948. We used LY294002 to rescue the abnormal GSK-3β. Results in our study showed that LY294002 slowed down the migration and strengthened the susceptibility of 4T1 cells to specific cytotoxicity, while promoting the activity of NK cells. Despite NK cells being only a fraction of immune microenvironment, the discovery makes sense to modify NK cells and improve the response of tumor cells to NK cells. Given that chimeric antigen receptor (CAR)-NK cell- and NK cell activating receptors (NKR)-NK cell-based immunotherapies are promising new strategies for cancer therapy49, and the promising results of this study lay foundation to accelerate the development and application of such immunotherapy.
A recent study showed that NOX2-derived ROS facilitated metastasis of melanoma cells by downregulating NK-cell function50, and our study illuminated that TWS119 induced a higher-level of ROS by upregulating the expression of NOX3 and NOX4 in 4T1 cells. Therefore, we deduced and confirmed that such accumulated ROS further inhibited the function of NK cells and promoted lung metastasis in breast cancer mice model. At this point, our result was in line with that of a previous study, which indicated that NOX4 was a potential target for intervention of cancer metastasis51. Our results in this study may provide a novel strategy for intervening breast cancer by combined application of adoptive NK cells and NOX4 inhibitor or pharmaceutical inhibitor of GSK-3β. However, the GSK-3β pharmaceutical inhibitor always resulted in a serious drug resistance as a clinical reaction. Therefore, to enhance the anticancer potential52, we should focus on attenuating pSer9-GSK-3β that may lead to drug resistance. Thus, NOX4 inhibitor is a better option, and must be studied further.
Besides, mitochondrial respiratory chain complexes are key sources of ROS generation and major regulators of cell apoptosis, among which complexes I and III are the most important53. Our study indicated that the activity of complexes I and III was positively correlated with the phosphorylation of GSK-3β at Ser9. Therefore, we can conclude mitochondrial respiratory chain complex is emerging as a novel target, and it could be regulated by pSer9-GSK-3β in 4T1 cells. Previous studies have shown that the activity of complexes was most remarkable in most aggressive breast cancer and Mito-TAM, a novel mitochondrial-targeted derivative of tamoxifen, and suppressed HER-2 breast cancer via selective disruption of respiratory supercomplexes54,55. Taking species differences and tumor heterogeneity into consideration, confirmation in human breast cancer tissue is still indispensable to get accurate conclusions.
In addition, we paid more attention to the phosphorylation of GSK-3β at Ser9, which inactivates it, while phosphorylation at Tyr216 site was not considered. Is there a balance in the regulation of the phosphorylation at Ser9 and Tyr216 sites? The problem has not been solved yet. Therefore, we have come to the conclusion that the inhibitor of GSK-3β promoted the growth and metastasis of mouse breast cancer, but more experiments are needed to be performed to elucidate whether the direct activation of GSK-3β can restore IFNγ-dependent, NK cell-mediated elimination of tumor cells.
Finally, eIF2B was identified to be regulated by ROS, which was induced by inactivated GSK-3β. In fact, GSK-3β regulates many downstream molecules by phosphorylating these substrates, some of which are tumor suppressors, such as phosphatase and tensin homolog deleted on chromosome ten (PTEN), and some of which are tumor promoters, such as C-myc. So GSK-3β affects breast cancer via a comprehensive interaction of different signals, and as a downstream molecule of PI3K/Akt/GSK-3β/ROS signaling pathway, among which eIF2B could be a major target. Since GSK-3β inactivated eIF2B by phosphorylating eIF2B-ε Ser53,56, and different studies have reported that ROS inactivated eIF2B by oxidative modification to the serine residue, overproduction of NAD+ and NADP+ caused by mitochondrial dysfunction could inhibit the activity of eIF2B57, and results from our study showed that dysfunctional eIF2B upregulated NKG2DLs, we concluded that GSK-3β/ROS/eIF2B plays an important role in regulation of the sensitivity of tumor cells to NK cells.
In summary, we demonstrated that GSK-3β, a tumor suppressor, could regulate the activity of NK cells and the susceptibility of tumor cells by controlling the level of ROS, and therefore, could modulate cancer growth and metastasis. Combined with our previous study, which showed that ROS promotes isoprenaline-induced M2 macrophage polarization29, the results in this study show ROS as a potential target for cancer therapy, and in particular, NOX4 treatment may be potentially used to disrupt breast cancer metastasis. It has been previously reported that γδT cells suppress cutaneous malignancy via NKG2D-associated regulation58; therefore, it deserves further attention. In addition, it must be further studied if GSK-3β/ROS plays a major role in T cell-mediated regulation of breast cancer, including γδT cells as well as integral T cell-related system.
The regulation of GSK-3β/ROS/NOX4 axis must be further studied to develop novel therapeutic strategies for breast cancer. EGFR/RAS/RAF/MEK/ERK/MARP signaling is common in all kind of cancers, and double inhibition by BRAF (darafenib) and MERK (trametinib) have been approved for the treatment of NSCLC, melanoma, and anaplastic thyroid cancer with BRAF V600E mutation by the FDA, based on the clinical trials for NCT01336634 and NCT02034110. Similarly, double inhibition by pSer9-GSK-3β and NOX or eIF2B may prove to be a viable alternative option.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 8117975 and 31770968) and Tianjin Institutes for Basic Sciences (Grant No. 15JCYBJC26900 and 16JCQNJC11700). We thank Professor Phillip Bryant (Nankai University) and Chang Liu (Tianjin University of Sport, China) for linguistic support.
Footnotes
↵*These authors contributed equally to this work.
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received August 13, 2018.
- Accepted November 1, 2018.
- Copyright: © 2019, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Glioma-Associated Oncogene Family Zinc Finger 2 (GLI2) Activates Wnt Signaling through Transcriptional Inhibition of Neuronal Precursor Cell-Expressed Developmentally Downregulated 4 (NEDD4L) to Promote Androgen-Induced Granulosa Cell Damage
- Bilirubin inhibits the anticancer activity of sorafenib by blocking MCL-1 degradation in hepatocellular carcinoma cells
- Ethyl {beta}-Carboline-3-Carboxylate Increases Cervical Cancer Cell Apoptosis Through ROS-p38 MAPK Signaling Pathway
- An ATF24 peptide-functionalized {beta}-elemene-nanostructured lipid carrier combined with cisplatin for bladder cancer treatment