

keywords
Introduction
Rhabdoid meningioma (RM) is a special type of meningioma. The pathologic features of RM include sheets of rhabdoid tumor cells with or without the findings typical of conventional meningiomas. Since the disease is rare, and the histologic structure similar to that of other tumors, misdiagnosis and failure to include the entity in the differentiation can occur from time to time. In this article, the pathologic features and immunophenotype of RM are analyzed, in combination with analysis of a case treated at our institution and a review of the literature.
Case Report
A male patient of 23 years visited our clinic with complaints of headache and blurred vision for 1 month. Physical examination showed a soft, palpable approximately 1 cm × 2 cm mass at the right temporoparietal area, and a partial skull defect. Visual examination was within normal limits, without visual field deficits or other abnormalities. MRI showed a right temporoparietal space-occupying lesion, with mixed signals of high and low density and a sharply defined border. A large area of associated peripheral edema was present a with midline shift towards the uninjured side and partial involvement of the cranial bone. No definitive meningeal tail sign (Fig. 1) was seen in the images. Surgical findings indicated that the tumor was 5 cm × 5 cm × 4 cm with a clearly defined border. The tumor was adherent to the overlying cranial bones. However, tumor infiltration into the cranial bones was not found, and the meningeal blood supply to the area was intact.

MRI: Slightly high intensity signals ranks first in T2W1, with a well defined border and obvious peripheral edema.
Following resection, the surgical specimen was fixed in 10% neutral formalin. Paraffin embedding, sectioning and H&E staining were performed, per standard procedure with subsequent microscopic examination. Immunohistochemical SP method was used to determine vim, EMA, GFAP, S-100, CK, NSE, HMB45, actin, desmin, Ki-67 and LCA. All reagents were obtained from Beijing Zhongshan Golden Bridge Biotechnology Co. Argentation was used for reticular fiber staining.
Histologic examination showed that the tumor was composed predominantly of striomuscular blastoma cells. The tumor cells formed a trabecular or diffusely lamellar pattern (Fig. 2), and were large, rounded with an irregular shape and abundant eosinophilic cytoplasm. Dysplasia was apparent in some cells, and giant and multinucleated giant tumor cells were found. The cell nuclei were round and, irregular in shape, and the nuclear chromatin was corpuscular lightly basophilic with focal areas of dark staining. Nucleoli were easily identified, ranging from 1 to 2 per nucleus. Nuclear inclusions and pathological karyokinesis could be seen in the cytoplasm (Fig. 3), with obvious focal necrosis. The tumor was rich with blood vessels, and focal mucinous change of the intercellular substance was observed (Fig. 4). A few areas with features typical of conventional meningiomas, including psammoma bodies, were present, located around the tumor margin.
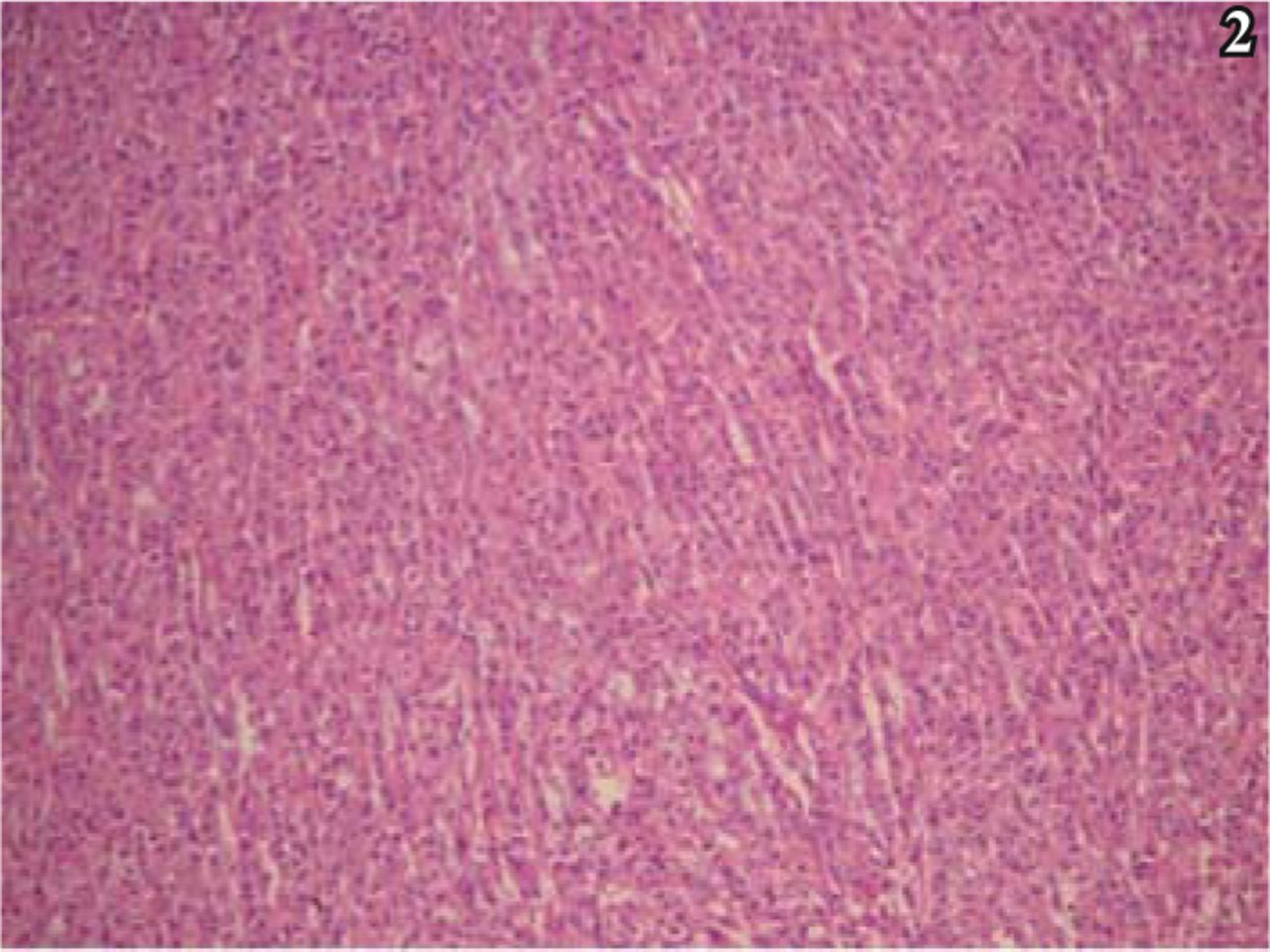
Trabecular or diffusely lamellar distribution of the tumor cells, with abundant eosiniphilic cytoplasm,; H&E staining, × 40.

High-power microscopy shows a heterogenity tumor cells, with a abnormal mitoses, and apparent intracytoplasmic and intranuclear inclusions, H&E staining, ×200.

Apparent mucinous change in local intercellular substance, H&E staining, × 200.
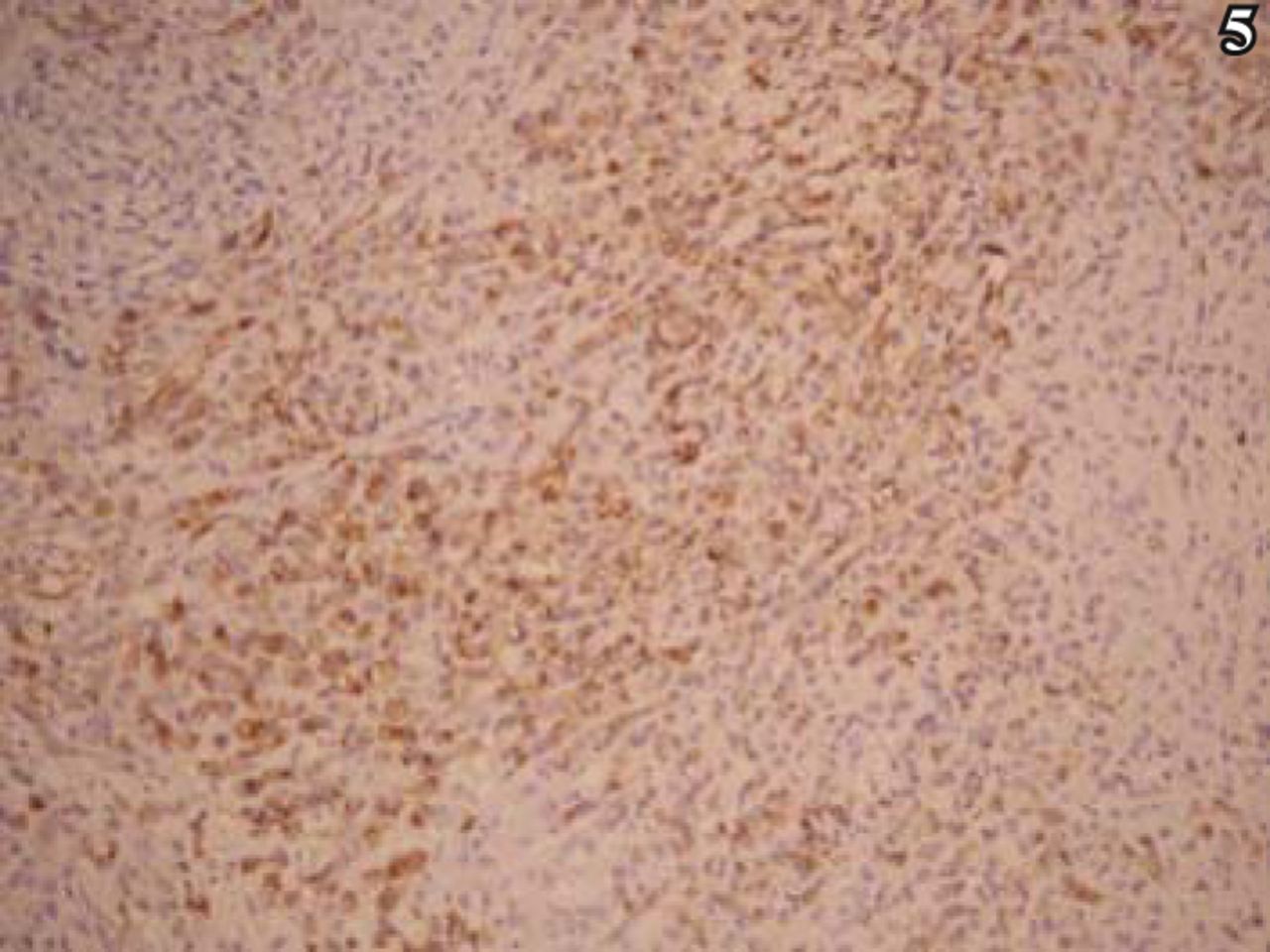
On immunohistochemical analysis, the tumor cells were diffusely and strongly positive for vim, EMA, and desmin (Figs. 5, 6). The cells were positive for GFAP in a lamellar pattern, and weakly positive for NSE and NF. In addition, the tumor cells were negative for CK, HMB45, S-100, actin, myoglobin, CgA, Syn, CD34, Mac387 and LCA. Positive rate of Ki-67 was partially > 10% (Fig. 7).

Positive expression of EMA, S-P method, ×100.

Strong positive expression of desmin, S-P method, × 100.

Ki-67 positive rate > 10%, S-P method, ×100.
The result of reticular fiber staining showed that the tumor was reticular fiber rich, in a trabecular distribution (Fig. 8).

Fascicular distribution of reticular fibers; argyrophilic staining, × 100.
The patient was given the pathologic diagnosis of RM of the right temporoparietal lobe, grade III, in accordance to the WHO classification.
Discussion
RM is an unusual tumor with high malignant potential. It has been reported overseas since 1991, and was first given the name by Perry et al.[1] in 1998. RM has been regarded as a kind of meningioma with a strong propensity for invasion. The existence of the rhabdoid cells in the tumor is a hallmark of this interesting neoplasm. In 2000, RM was classified as a new subtype of meningioma by the Nervous System Classification of WHO, and was graded as Stage III in the WHO classification of tumors. To date, dozens of cases of RM have been reported overseas[1-5] and about 10 cases have been reported in China[4-6]. Correct pathologic diagnosis is significant to clinical practice since the disease is rare, has a high malignant potential and poor prognosis. On review of the present clinical data, we found that RM may occur in all age groups, ranging from 5 to 84 years in our analysis, with a majority in middle-aged and young patients. However, compared to the age of patients with the much more common conventional meningioma, the onset age of RM patients is relatively low[5]. There appeared to be no significant difference between RMs and common meningiomas in the male: female ratio of incidence, in the pathogenic site or in the clinical symptoms. Nevertheless, the postoperative relapse rate of RM approaches 87% and has a 3-year survival time and a very poor prognosis. MRI and CT usually indicate an intracranial space-occupying lesion, with a sharply defined border, heterogenous signals, and obvious peripheral edema.
The pathologic features of RM are as follows. The gross appearance of RM is similar to that of the common meningioma, but RM is quite soft in texture. The cut surface is grey-red to grey-yellow, with secondary changes including necrosis and hemorrhage. Microscopically, RMs are composed of sheets of rhabdoid tumor cells and typical meningothelial cells in varying proportions. The rhabdoid tumor cells are usually present in a trabecular, false-acinar, or diffusely lamellar alignment, and RMs with a papillary pattern are infrequently seen. Tumor giant cells, polykaryocytes, abnormal mitosis, necrosis, bleeding, and blood vessel hyperplasia are easily found. Electron microscopic visualization indicates that there are few simple membranous connections between the tumor cells, and that the cells lack myofibrillar structures, such as mosaicism (inlaid connection), myofilament, and Z-belt.
In regard to the immunophenotype of RM, authors worldwide agree on the consistent expression of vimentin and EMA in RM, but their viewpoints on muscle-derived markers vary greatly. All of the muscle-derived markers in the RM cases reported in China have been negative, while these markers are reported as positive in cases from overseas. The muscle-derived markers typically include desmin, actin and SMA[3]. CK and S-100 can be either positive or negative[4,5]. Interestingly, Bergmann et al.[3] found expression of proteins, such as GFAP, NSE, and HSP70 in RM. In our case, the expression of vim, EMA and desmin in RM cells showed strong and diffuse positivity. The tumor cells were also positive for GFAP, staining in a lamellar pattern. These results are consistent with similar cases reported in the literature overseas.
Concerning the differential diagnosis, although the architectural features of conventional meningiomas are present in most cases of RM, the diagnosis between RM and meningioma is relatively easy, when the pathogenic site and the immunological marker profile is considered. However, if the syncytial pattern characteristic of common meningiomas is absent, misdiagnosis may occur as follows. i) Melanoma-the cytoplasm of RM cells is rich and strongly eosinophilic, and entoblast can be easily seen. The RM cells are arranged in a trabecular to adenomatiod pattern, similar to malignant melanoma (MM). In some cases it can be especially difficult to discriminate RM from MM as MMs can have a rhabdoid component. However, immunodetection by HMB45, indicative of melanin, in combination of with lack of staining for desmin and SMA, can distinguish between these two neoplasms. ii) Atypical teratoma and rhabdoid tumor (AT/RT)-morphologically similar rhabdoid cells can be found both in AT/RT and in RM. Immunological tumor markers, such as vimentin, EMA, GFAP, desmin, and SMA can all be positive in RM and AT/RT, increasing the difficulty in differentiating RM from the latter. However, AT/RT is found mainly in the children under the age of 5 years with a predilection for the rear-cranial fossa. Thus, AT/RT does not involve the meninges. Further neuroectodermal, epithelial or mesenchymal components are mixed in most of these tumors. In addition, studies have shown that absence of 22q and inactivity of IN1 often occurs in AT/RT. Although there is also an absence of 22q in RM, inactivity of INI1 is rare. Therefore, detection of INI1 activity could be used to differentiate RM from AT/RT[7]. iii) Megacell medulloblastoma (MMB)-the shape of tumor cells in MMB is similar to that of the RM cells. However, MMBs most frequently occur in children, and are typically located in the vermis cerebellum, therefore, the lack of involvement of the meninges can further distinguish it from RMs. The immunological profile of MMBs is GFAP, NF and Syn, positive, and EMA and desmin negative; these features can also be used for identification. In addition, it is necessary to further discriminate RM with mucinous change, from chordocarcinoma-like meningioma, and chordocarcinoma.
- Received May 11, 2009.
- Accepted July 2, 2009.
- Copyright © 2009 by Tianjin Medical University Cancer Institute & Hospital and Springer
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.