

Abstract
Ovarian cancer is the second most lethal gynecological cancer worldwide and while most patients respond to initial therapy, they often relapse with resistant disease. Human epidermal growth factor receptors (especially HER1/EGFR and HER2/ERBB2) are involved in disease progression; hence, strategies to inhibit their action could prove advantageous in ovarian cancer patients, especially in patients resistant to first line therapy. Monoclonal antibodies and tyrosine kinase inhibitors are two classes of drugs that act on these receptors. They have demonstrated valuable antitumor activity in multiple cancers and their possible use in ovarian cancer continues to be studied. In this review, we discuss the human epidermal growth factor receptor family; review emerging clinical studies on monoclonal antibodies and tyrosine kinase inhibitors targeting these receptors in ovarian cancer patients; and propose future research possibilities in this area.
keywords
Introduction
Ovarian cancer is the eighth most lethal type of cancer in women worldwide, with over 184,000 fatalities reported in 20181. It has a poor prognosis and is usually diagnosed at late stage, due to lack of specific diagnostic biomarkers and relatively non-specific symptoms. Current diagnostic tests for ovarian cancer include the CA-125 blood test coupled with abdominal/pelvic ultrasound and computerized tomography (CT) scan. First line treatment of ovarian cancer is optimal debulking of macroscopic disease, generally followed by chemotherapy with carboplatin alone or in combination with paclitaxel2,3.
The most common type of ovarian cancer is epithelial ovarian cancer (EOC), which is divided into 5 main subtypes, with differing histological, molecular, and genetic characteristics3,4. The most common subtype is high grade serous ovarian cancer (HGSOC) which accounts for approximately 70% of the cases, with clear cell (10%), endometrioid (10%), mucinous (< 5%), and low grade serous cancer (< 5%) comprising the other significant subtypes3,4. Recent investigations into the pathogenesis of ovarian cancer showed that it primarily originates from different parts of the female reproductive system and involves cellular migration to the ovaries. HGSOC is identified to originate from the distal fallopian tube, endometrioid and clear cell cancers arise from the endometrium, while low grade serous cancer might progress from serous cystadenoma and serous borderline tumors5.
Multiple drugs have been tested and approved for ovarian cancer although the response rate for second line therapy is only 10%–35% and different ovarian cancer subtypes respond differently to drug treatment. HGSOC patients usually respond well to initial platinum-based therapy, given their BRCA and p53 mutations7. However, these patients often present with resistance to initial therapy after a few months.
There is increasing interest in the potential use of targeted inhibitors for the treatment of ovarian cancer. This review seeks to overview the current clinical and preclinical status of human epidermal growth factor receptor (HER) targeted therapy in ovarian cancer, with special emphasis on tyrosine kinase inhibitors (TKIs).
Human epidermal growth factor receptors (HERs)
The HER family has been associated with the progression of several cancers including breast, lung and colon cancer8. In ovarian cancer, amplification, and/or high expression of epidermal growth factor receptor (EGFR), HER2 and HER3 receptors have been implicated in the progression and prognosis of the disease9-12.
The HER family of receptors [also known as erythroblastic leukemia viral oncogene (erbB) family] are present on the cell surface as monomers, in the absence of ligand activation. There are four members in this family, EGFR (HER1/erbB1), HER2 (neu/erbB2), HER3 (erbB3) and HER4 (erbB4) (Figure 1). With the exception of HER2, ligands bind to their extracellular domain and form homo- or heterodimers with other members of the family, preferentially with HER2, since it has the most favorable kinase activity and exists in an activated form13. HER ligands are divided into three groups; those which bind specifically to EGFR (epidermal growth factor, amphiregulin and transforming growth factor-α), those conferring dual specificity to EGFR and HER4 (betacellulin, heparin-binding EGF, and epiregulin), and those which bind to HER3 and HER4 (neuregulins/heregulins)14.

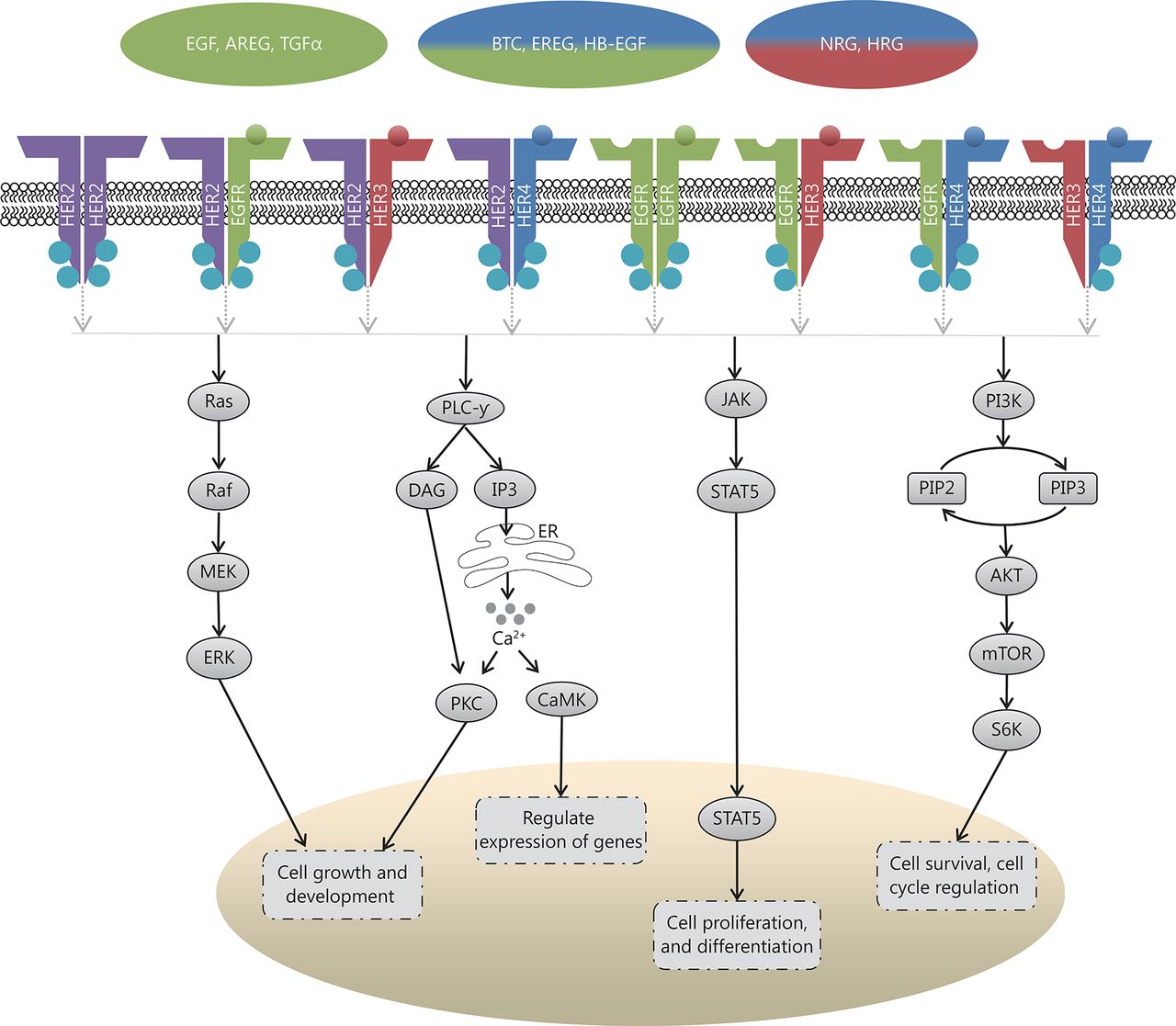
Scheme illustrating the downstream signaling of the HER pathway, chiefly the mitogen activated protein kinase (MAPK)/ERK pathway, the phospholipase Cγ (PLCγ) pathway, the signal transducer and activation of transcription (STAT) pathway, and the phosphoinosidyl-3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway; where ER=Endoplasmic Reticulum, Ca2+=Calcium ions
Upon receptor dimerization, multiple downstream pathways are activated, which regulate cell proliferation, differentiation, angiogenesis, survival, and cellular metabolism amongst other functions. Heterodimerization allows for a myriad of phosphotyrosine residues to bind, which in turn increases the possibilities for signaling pathways15. These pathways (Figure 1) include the mitogen activated protein kinase (MAPK)/ERK pathway, which regulates the growth and development of cells, the signal transducer and activation of transcription (STAT) pathway, which governs cell proliferation and differentiation, the phosphoinosidyl-3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway, which regulates cell survival and metabolism, and the phospholipase Cγ (PLCγ) pathway, which controls calcium-dependent actions13,16,17. In tumorigenesis, mutations within the components of these pathways can cause cancer cells to acquire certain aptitudes, including impartiality to proliferation signals, circumvention of apoptosis, insensitivity to growth inhibitory signals, augmented replicative potential and the capability to metastasize18.
EGFR (HER1) is a receptor tyrosine kinase comprising an extracellular ligand-binding domain of 622 residues, a 23 residue transmembrane domain, and a large, 522-residue intracellular domain15. EGFR is normally weakly expressed in the ovaries, however, several studies have found that EGFR is highly expressed in ovarian cancer. Immunohistochemical studies have indicated that 30%–70% of ovarian cancers have increased EGFR expression19-21. High expression of EGFR is associated with poor, progression-free survival (PFS), advanced tumor grade, greater residual tumor mass and rapid proliferation9,10,12. It has also been suggested that high EGFR expression in the tumor stroma is associated with aggressive clinical conditions and outcome and EGFR upregulation in fibroblasts is associated with growth and migratory abilities of ovarian cancer cells22.
HER2 is overexpressed in approximately 6%–30% of ovarian cancer patients10,23 and is initially associated with DNA amplification and poor prognosis24. Overexpression is often detected in the mucinous (19%)25 and clear cell (14%)26 subtypes. However, even some serous (3%) and endometrioid ovarian cancers (2%) have HER2 overexpression27. Several studies have associated overexpression of HER2 with poor prognosis10,28.
HER3 is expressed as a full-length receptor on the cell surface, in parallel with truncated intracellular isoforms. However, the activity of the latter is not well defined29,30. HER3 lacks tyrosine kinase activity, hence it has to be transphosphorylated by other HER members to promote cell signaling15. HER3 is more frequently expressed in ovarian cancer (30%–80%) than EGFR and HER231 and is more common amongst borderline and early-stage lesions32. Among the dimerization possibilities between these proteins, the most potent signaling complex is generated when HER3 heterodimerizes with HER233. Increased HER3 expression has been associated with poor clinical outcome and the average survival time for patients with low HER3 expression was 3.3 years, in contrast to 1.8 years for patients with high HER3 expression11. Studies in various cancers have shown that when HER3 and MET are co-expressed, they are often associated with either response or resistance to therapy34-36. Other studies have shown that high expression of HER3 might lead to HER3-PI3K-Akt signaling cascade in doxorubicin and cisplatin treated ovarian cancer, which often results in resistance to therapy37,38.
HER4 is the least understood receptor of the HER family. It occurs as a spliced isoform, often being processed further by enzymes into a soluble intracellular domain, which can disperse to the cell cytoplasm or nucleus39. In breast cancer, nuclear localization of the intracellular domain in combination with estrogen expression predicted worse clinical outcomes compared to membrane HER4 and estrogen40. There are conflicting views about the expression of HER4 in ovarian cancer, with earlier reports suggesting either decreased or lack of expression of the receptor41, while more recent studies suggest an increased expression of HER4 in malignant tissues compared to normal tissues31,42,43. Although the implication of HER4 expression in ovarian cancer is unclear, two studies found a possible correlation between HER4 expression and resistance of serous ovarian cancer to chemotherapy42,44.
Monoclonal antibodies
HER-targeted monoclonal antibodies (mABs), such as trastuzumab (Herceptin®) and pertuzumab (Perjeta®) are recombinant humanized mABs, inhibiting HER2 extracellularly with differing modes of action (Figure 2). These agents have shown favorable results in HER2 positive cancers, especially HER2-positive breast cancer, where they are well established as standard therapy. More recently, trastuzumab-emtansine antibody-drug conjugate has been developed as another option for trastuzumab-resistant disease. Preclinical in vitro and in vivo studies and clinical trials have been focusing on the activity of these mABs in ovarian cancer, especially in selective subtypes, particularly mucinous cancers, which have HER2 amplification and overexpression45,46.
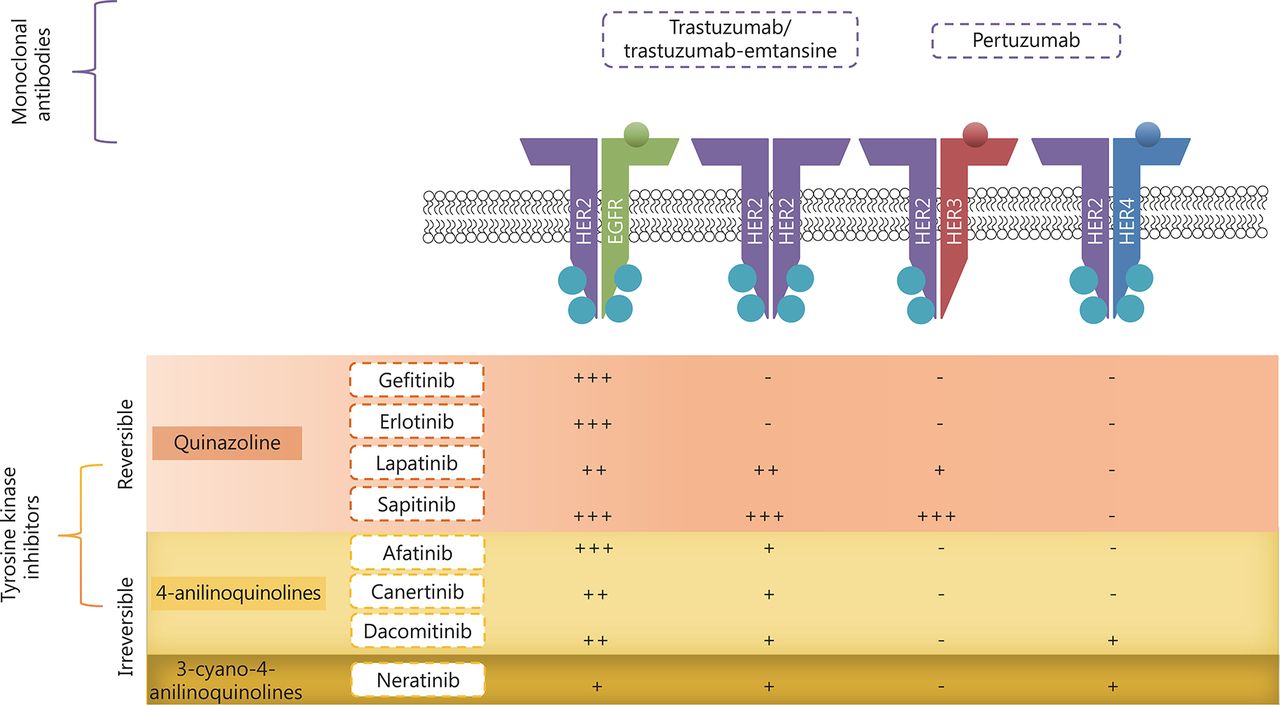
Site of action of the various TKIs and the relative potency towards the receptor; where +++ is very potent, ++ is potent, + is mildly potent and – is generally not active.
Trastuzumab
Trastuzumab binds to the juxtamembrane region of the extracellular domain of HER2, inhibiting cleavage of the extracellular domain, obstructing HER2 homodimerization as well as inducing antibody dependent cell-mediated cytotoxicity47,48. Trastuzumab requires HER2 overexpression for it to be effective48,49. The success of trastuzumab in clinical trials49-51 has led to its clinical approval in metastatic breast cancer overexpressing HER2, as monotherapy or in combination with paclitaxel depending on whether the patients previously received chemotherapy regimens for their metastatic disease52. A phase II clinical trial of trastuzumab involving 41 eligible ovarian cancer patients with HER2 overexpression demonstrated an overall response rate (ORR) of 7.3%, which included one complete and two partial responses. The median PFS was two months53.
Pertuzumab
Pertuzumab acts by blocking the dimerization domain of HER2, thus inhibiting HER2 heterodimerization48. Unlike trastuzumab, it does not require HER2 overexpression to confer its inhibitory effects54. Pertuzumab is used to treat HER2-positive metastatic breast cancer patients who have not been previously exposed to anti-HER2 therapy or chemotherapy for metastatic disease and is also used for the neoadjuvant treatment of HER2-positive early stage breast cancer55. In a phase III clinical trial for breast cancer (CLEOPATRA), the combination of trastuzumab and pertuzumab along with docletaxel, showed additional benefit compared to monotherapy, which has subsequently led to the approval of this combination for HER2-positive metastatic breast cancer56. A randomized phase II clinical trial with pertuzumab showed better PFS (5.3 months) in low HER3 expressed, platinum-resistant ovarian cancer patients and hence it was assessed in a phase III clinical trial (PENELOPE) in platinum-resistant ovarian cancer patients with low expression of HER357,58. In the PENELOPE trial, patients who received pertuzumab with chemotherapy showed a PFS of 4.3 months and an ORR of 13.1%, when compared to the PFS of 2.6 months and ORR of 8.7% in patients who received placebo with chemotherapy58. In an in vivo setting using ovarian cancer xenograft mouse models, our group has demonstrated that the combination of pertuzumab and trastuzumab produces prolonged growth inhibition, when compared to either antibody used as a single agent59. Furthermore, another preclinical study has suggested that trastuzumab could also augment sensitivity to endocrine therapy in ERα-positive ovarian cancer60.
Trastuzumab-emtansine
Trastuzumab-emtansine (T-DM1, Kadcyla®), is a HER2 targeted mAB conjugated to a microtubule inhibitor (emtansine). T-DM1 acts by binding to HER2, triggering the endocytosis of the HER2-T-DM1 complex. Once in the cytoplasm, DM1 is released from the complex, which inhibits microtubule assembly, leading to cell death61. In a phase III clinical trial for breast cancer, T-DM1 treated cohort showed a superior clinical outcome (PFS 9.6 months) compared to patients who received lapatinib with capecitabine (6.4 months)62. It was later approved as monotherapy for HER2-positive metastatic breast cancer, which is resistant to trastuzumab treatment63. In a recent in vivo study by Menderes et al.64, T-DM1 demonstrated significant tumor growth inhibition against HER2 overexpressing ovarian cancer primary cell lines compared to either pertuzumab or trastuzumab alone or a combination of both. It also reduced tumor growth and improved survival in xenograft models64-66. This agent was also shown to have similar antibody-dependent, cell-mediated cytotoxicity as pertuzumab, trastuzumab and their combination64. Another study demonstrated their excellent inhibitory activity against both subcutaneous and intraperitoneal growth of the SKOV3 in an ovarian cancer xenograft model67.
TKIs
TKIs are small drug molecules that inhibit tyrosine kinases. Tyrosine kinases include the HER family, vascular endothelial growth factor receptors (VEGF), platelet-derived growth factor receptors (PDGFR), and also non-receptor tyrosine kinases BCR-ABL and KIT68,69. Tyrosine kinases are enzymes that catalyze the transfer of phosphate from adenosine triphosphate (ATP) onto target proteins to elicit a response. There are three types of TKIs. Most small molecule TKIs are type I, which compete with ATP by binding to ATP binding sites on the active conformation of the receptors, thereby interfering with the action of tyrosine kinases69. Type II TKIs bind to the inactive conformation of a kinase, while type III allosteric inhibitors bind to sites distant from the active site70. To date, a few TKIs have been evaluated in ovarian cancer patients that are described below (Table 1, Figure 2). These include the first generation EGFR inhibitors, gefitinib and erlotinib, which have shown clinical efficacy against mutant EGFR lung cancer. Since, a resistance mutation develops frequently at T790M upon treatment, covalent irreversible second generation TKIs were developed. These consist of afatinib and neratinib that are active against this mutation. Other HER inhibitors were developed with broader inhibitory activity across multiple HER family members (pan-inhibitors) and these include lapatinib and canertinib as early developed inhibitors, followed by neratinib, sapitinib, and dacomitinib. Finally, multi-targeted TKIs that target the HER family among other targets (e.g., PDGFR, VEGFR, etc.) include vandetanib and leflunomide.
HER-targeted TKIs evaluated in preclinical models of ovarian cancer
Reversible inhibitors
Gefitinib
Gefitinib (Iressa®, ZD-1839) is a type I quinazoline derivative TKI, reversibly selective for EGFR. Gefitinib is marketed for monotherapy of locally advanced or metastatic NSCLC and is most effective in cancers with mutation or increased EGFR copy number81. When evaluated in an ovarian cancer phase II trial, gefitinib showed a limited response rate in platinum pre-treated ovarian cancer patients, with only one out of twenty-seven patients having an ORR of 4%82. However, this cancer was the one in this cohort harboring a mutation in the catalytic region of EGFR, consistent with data for NSCLC. Another phase II clinical trial combining gefitinib with tamoxifen in refractory or resistant ovarian cancer patients, did not demonstrate any advantageous tumor responses with median time-to-progression being 58 days83. Preclinical studies in ovarian cancer demonstrate that combining gefitinib with cisplatin, increases the efficacy of cisplatin, mainly due to the inhibition of downstream EGFR signaling and blocking DNA repair mechanisms84.
Erlotinib
Erlotinib (Tarceva®, OSI-774) is a quinazoline derivative, which reversibly inhibits EGFR. It is indicated as a first line therapy for NSCLC and in combination with gemcitabine for pancreatic cancer. Erlotinib has shown enhancement of PFS compared to chemotherapy (13.1 months vs. 4.6 months, respectively) in chemotherapy-naive NSCLC patients, harboring EGFR mutations85. An in vivo study in ovarian cancer expressing high EGFR demonstrated that a combination of erlotinib with olaparib, a PARP inhibitor, had a greater tumor suppressive effect than monotherapy86. A phase II study combining erlotinib with carboplatin showed that this combination was more effective in ovarian cancer patients with platinum-sensitive disease compared to platinum-resistant disease with 57% and 7% objective response rates, respectively. However, the contribution of erlotinib in this combination is unclear87. Another phase II study assessing the pathologic complete response (pCR) of ovarian cancer patients administered with a combination of carboplatin, paclitaxel, and erlotinib, resulted in pCR of around 30%, which was not an improvement when compared to previous results88. Continuous infusion of topotecan with erlotinib was studied in a phase II trial, where only 1 out of 6 patients showed a satisfactory partial response89. An exploratory phase II clinical trial involving bevacizumab and erlotinib determined that high levels of VEGF-A caused bevacizumab resistance, while erlotinib did not seem to contribute to the efficacy of the combination90. A randomized phase III study evaluated the efficacy of administering erlotinib to patients with ovarian cancer after first line chemotherapy. The median PFS in patients receiving erlotinib and placebo was 12.7 and 12.4 months, respectively, concluding that erlotinib does not render pre-treated ovarian cancer patients with additional benefits91. In extension to this phase III clinical trial, a tissue biomarker study concluded that increased EGFR gene copy number led to worse overall survival and PFS92.
Lapatinib
Lapatinib (Tyverb®, GW-572016) is an oral competitive TKI inhibitor selective for EGFR and HER2. It is recommended in HER2 overexpressing breast cancer. Preclinical data indicates that lapatinib is effective when HER2 is overexpressed and most likely homodimerized78. In breast cancer clinical trials, lapatinib has shown better inhibition of tumors expressing HER2 instead of EGFR78,93. In a phase I study in ovarian cancer, while assessing lapatinib in combination with carboplatin many non-dose limiting toxicities were noticed and 6 out of 11 patients had PR or stable disease94, while in a phase II trial (LapTop) assessing lapatinib with topotecan, only 20% of patients experienced benefit. However, considerable hematologic adverse effects were observed in this trial95,96. In another phase II study in recurrent ovarian cancer, the median PFS was 1.8 months, OS was 10.5 months, and only 2 out of 25 patients had PFS at 6 months, while there was no ORR, which might be due to low EGFR and HER2 expression97.
Sapitinib
Sapitinib (AZD 8931) is a type I, reversible, equipotent inhibitor of EGFR, HER2 and HER3 receptor signaling, especially when EGFR is highly expressed and there is no HER2 overexpression78,98. In fact, sapitinib has shown enhanced tumor growth inhibition against EGFR-driven xenograft tumors when compared to lapatinib78. Preclinical studies suggest that sapitinib favors HRG-induced HER2/HER3 heterodimers78. It has also demonstrated its ability to inhibit proliferation through pERK and pAkt pathways, and induce apoptosis through M30 and cleaved caspase-378. In xenograft models of inflammatory breast cancer, sapitinib alone significantly inhibited tumor growth, however, the combination of paclitaxel and sapitinib was more effective than either agent alone98. Sapitinib has been tested in 8 ovarian cancer patients amongst others, in a phase I clinical trial, to assess the maximum tolerated dose, which was established to be 240 mg twice, daily99.
Irreversible inhibitors
Afatinib
Afatinib (Gilotrif®, Giotrif®, BIBW-2992) is a type I anilinoquinazoline derivative TKI inhibitor, which irreversibly binds to EGFR, HER2 and HER4100. It is currently approved for the treatment of mutated EGFR non-small cell lung cancer (NSCLC). Afatinib has not been extensively tested in ovarian cancer clinical trials. A phase I dose escalation study, which included four ovarian cancer patients, showed promise clinically as indicated by stable disease101,102. In breast cancer, afatinib treatment was effective, however, it produced many unwanted side effects in patients103,104. In an ovarian cancer preclinical study, afatinib reversed the ATP binding cassette (ABC) mediated multidrug resistance to paclitaxel and adriamycin and also increased the apoptotic efficacy of paclitaxel in ABCB1 overexpressing tumors105. In breast cancer, it was also found that afatinib overcomes HERT798I-mediated neratinib resistance106. In vivo, afatinib with docetaxel showed better response in tumor size reduction, than either drug as a single agent107. In vitro studies using ovarian cancer cell line models showed that afatinib is effective in inhibiting migration and proliferation108. It was also found to be effective in inhibiting basal and heregulin-induced EGFR, HER2, Akt and ERK phosphorylation108.
Canertinib
Canertinib (CI-1033, PD-183805) is a 4-anilinoquinazoline, irreversible, pan-HER TKI, which reached phase II clinical trials. However, it was recently withdrawn109. In vivo, canertinib showed potent inhibitory effects in ovarian cancer cell lines, especially when combined with a c-MET inhibitor (PHA665752), which further reduced phosphorylation and total expression of signaling proteins108,110,111. A randomized phase II clinical trial in platinum resistant or refractory ovarian cancer patients resulted in disease stability in about 30% of the patients and a one-year survival rate of around 37%. However, there were no complete or partial responses112. Studies suggest that high levels of HER and low levels of HER autocrine ligands lead to canertinib resistance113.
Neratinib
Neratinib (Nerlynx®, HKI-272) is an oral, irreversible pan-HER inhibitor, which has been recently approved for the adjuvant treatment of early stage HER2-positive breast cancer114, after a phase III study indicated a 2-year, invasive, disease-free survival rate of 94%, when administered after chemotherapy and trastuzumab adjuvant therapy115. In a phase II study assessing neratinib in advanced NSCLC, patients with T790M EGFR mutation did not respond to therapy, however, partial response or disease stabilization was seen in patients with G719X mutated EGFR116. Overall, previously treated patients or TKI-naïve patients did not benefit notably from neratinib treatment. One of the reasons for this could be low bioavailability due to dose reductions prompted by toxicity116. In HER2-positive breast cancer, neratinib as a single agent was well tolerated and has shown substantial clinical activity in trastuzumab-naïve patients, with a 16-week PFS rate of 78% and median PFS of 39.6 weeks versus 16-week PFS rate of 59% and median PFS of 22.3 weeks in trastuzumab pre-treated patients79. Other clinical trials in breast cancer assessed neratinib in combination with temsirolimus, vinorelbine, paclitaxel with or without trastuzumab and capecitabine, all of which were well tolerated by patients and had anti-tumor properties117-121. Neratinib has demonstrated pre-clinical efficacy in ovarian cancer, especially in HER2-amplified carcinosarcoma, where it inhibits proliferation and tumor growth122, as well as decreases phosphorylation of transcription factor S6 and causes cell cycle arrest in the G0/G1 phase123. There is currently a phase II clinical trial assessing neratinib efficacy in HER2-positive solid tumors (SUMMIT Trial) with mutations in EGFR, HER2 or HER3, including ovarian cancer (NCT01953926). This trial included four evaluable ovarian cancer patients of whom one had stable disease, while three had disease progression124.
Other HER-targeted TKIs investigated in clinical trials
Dacomitinib (PF00299804), a recently developed irreversible pan-HER receptor inhibitor, has demonstrated interesting anti-proliferative activity against chemoresistant ovarian cancer cell lines125. One ovarian cancer patient showed response in a phase I clinical trial of dacomitinib in combination with anti-IGFIR antibody figitumumab126. Several multi-targeted TKIs with broad-spectrum activity including the HER family are undergoing evaluation. Vandetanib (ZD6474) is a drug that inhibits EGFR, VEGF receptor and Ret signaling and has been tested as monotherapy in an ovarian cancer trial where, despite decreasing EGFR phosphorylation, it demonstrated little efficacy127. Leflunomide is an inhibitor of EGFR, PDGFR, and FGFR and in two phase II trials, 1 out of 8 (12.5%) and 1 out of 15 (7%) ovarian cancer patients demonstrated partial response128,129.
Biomarkers of sensitivity and resistance to TKIs in ovarian cancer
In breast and lung cancers, informative biomarkers of sensitivity to HER TKIs include overexpression of HER2 and mutation of EGFR. In breast cancer, HER2 overexpression is an effective biomarker of sensitivity to HER2-targeted TKIs, such as lapatinib. Preclinical studies of T-DM1 in ovarian cancers suggest that minimal expression of HER2 was essential for anti-tumorigenic properties of T-DM1 in model systems66. Analysis of a series of ovarian cancer xenograft models demonstrated the curative potential of trastuzumab/pertuzumab combination in cancers with amplification and overexpression of HER2. Currently there is less information available regarding the association of HER2 expression levels and TKIs, in ovarian cancer59,60. Interestingly, there is one case report of dramatic remission of a chemotherapy-resistant ovarian cancer to trastuzumab, which was HER2-negative suggesting that, the factors governing responsiveness in ovarian cancer might differ from those in breast cancer130. The mutated form of EGFR with deletions in exon 19 indicates sensitivity to TKIs such as erlotinib and gefitinib, in NSCLC131. However, the importance of EGFR mutations in ovarian cancer is still not well researched since the occurrence of these mutations is much lower. As mentioned above, in a phase II trial of gefitinib in 27 ovarian cancer patients, the single patient showing response did contain an EGFR mutation (2235del15; E746-A750del) in the catalytic domain consistent with this molecular feature being an indicator of sensitivity82. This requires further validation in future studies.
The importance of mutations in HER2 and HER3 for sensitivity to pan-HER inhibitors, is under clinical investigation at present and a basket trial (SUMMIT) investigating neratinib treatment in multiple cancers with mutations, has been reported124.
As observed with other chemotherapeutic drugs, resistance to TKIs is inevitable132. Mechanisms of resistance to EGFR-specific TKIs can include abnormalities in HERs, such as HER2 overexpression and mutations like EGFRvIII and HER2L869R106,133 and secondary EGFR mutations in T790M134, L747S, D761Y, and T854A135. Downstream signaling pathways that are frequently modified include mutations in KRAS, BRAF, PIK3CA, and PTEN136-138. Alternative pathways that can bypass control include aberrant activation of MET and HGF139,140, modifications in VEGF receptors which trigger vascular permeability, in platelet-derived growth factors that regulate angiogenesis and in interleukin-6 that controls inflammatory processes132-134. For other TKIs, overexpression of ABC resulting in low drug concentration in cells due to decreased uptake and increased efflux of the drug141 also contribute in resistance to therapy.
Future research
Combination therapy
Research in various cancers show that the combination of TKIs with chemotherapy, radiation, or mABs significantly inhibits tumor growth, without additional toxic effects, since they have different inhibitory profiles. For instance, lapatinib is administered in combination with either capecitabine/trastuzumab/aromatase inhibitor in HER2-positive breast cancers. Synergistic drug combinations can be achieved in two ways: vertically, which involves similar doses as monotherapy, and horizontally, in which the concentration of the dose is decreased downwards142. To date, in ovarian cancer, there are no established combination strategies involving TKIs. In ovarian cancer, only the mAB bevacizumab is approved to be used in combination with paclitaxel and carboplatin. In a phase II ovarian cancer study, the combination of pertuzumab and gemcitabine showed improved overall PFS when compared to gemcitabine and placebo57. In breast cancer, the combination of pertuzumab and trastuzumab has shown significant advantages over monotherapy. However, the combination of these mABs in ovarian cancer has only been studied in vivo, which has shown promising results59, and thus might be worth looking at clinically. Additionally, the combination of mABs and TKIs has not been clinically studied so far, in ovarian cancer. The combination of HER-targeted inhibitors with Poly (ADP-ribose) polymerase (PARP) inhibitors might prove useful in the treatment of ovarian cancer, since, recent in vitro studies combining a TKI with a PARPi showed synergistic growth inhibitory effects143. Computational biology has developed in recent years and offers the potential of precision medicine. Molecular anomalies can now be detected when screening through molecular information, which could identify individual patient appropriate medication . Hence, more pre-clinical bioinformatics studies need be conducted to investigate the effects of therapy on a molecular basis.
Chemosensitive versus chemoresistant phenotypes
HGSOC usually responds well to initial ovarian cancer therapy, with response rates as high as 85%7. This is frequently due to the fact that HGSOC has BRCA mutations. First-line chemotherapy acts by damaging DNA strands; BRCA acts to repair DNA, however, given that HGSOC frequently lacks BRCA function, DNA strands cannot be repaired, which consequently leads to the efficacy of primary chemotherapy in HGSOC. However, HGSOC cells through further mutation can restore BRCA function, which is one of the leading causes of resistance to initial chemotherapy144.
It is often acknowledged that ABC transporters play a pivotal role in resistance to first line chemotherapy. In fact, ABCB1, ABCB4, and ABCG2 were significantly up-regulated in cisplatin and paclitaxel resistant ovarian cancer cells145. Pertuzumab in a clinical trial of chemoresistant disease showed that the mAB demonstrated possible anti-tumor activity when combined with either gemcitabine or paclitaxel57,58, while novel TKIs that target multiple sites often demonstrate their ability to reverse ABC-mediated drug resistance105,146-148. Hence, mABs and TKIs might be more active in chemoresistant ovarian cancer than in the chemosensitive type.
Conclusions
Ovarian cancer is a complex disease, with multiple molecular profiles. It frequently becomes resistant after initial therapy necessitating the development of new strategies.
The use of HER-targeted therapy continues to be assessed in this disease, since it might have value for selective patients and pre-clinical data supports the potential of this approach. Only a limited number of phase II trials have been completed in ovarian cancer and while response rates are low, there are frequent good percentages of stable disease. The pan-HER TKIs may have broader efficacy and utility than the early EGFR-targeted TKIs, which are dependent on the presence of mutations (are uncommon in ovarian cancer). Further biomarker studies are now required to help identify the most sensitive ovarian cancers and combination strategies require further development.
Acknowledgements
The work disclosed in this publication is partially funded by the Endeavour Scholarship Scheme (Malta). Scholarships are part-financed by the European Union–European Social Fund (ESF)–Operational Program II–Cohesion Policy 2014–2020 “Investing in human capital to create more opportunities and promote the well-being of society”.
Footnotes
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received April 6, 2018.
- Accepted October 26, 2018.
- Copyright: © 2018, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.
- 72.
- 73.
- 74.
- 75.
- 76.
- 77.
- 78.↵
- 79.↵
- 80.
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.
- 119.
- 120.
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.
- 148.↵
In this issue






{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.