

Abstract
The intricate interplay between the human immune system and cancer development underscores the central role of immunotherapy in cancer treatment. Within this landscape, the innate immune system, a critical sentinel protecting against tumor incursion, is a key player. The cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING) pathway has been found to be a linchpin of innate immunity: activation of this signaling pathway orchestrates the production of type I interferon (IFN-α/β), thus fostering the maturation, differentiation, and mobilization of immune effectors in the tumor microenvironment. Furthermore, STING activation facilitates the release and presentation of tumor antigens, and therefore is an attractive target for cancer immunotherapy. Current strategies to activate the STING pathway, including use of pharmacological agonists, have made substantial advancements, particularly when combined with immune checkpoint inhibitors. These approaches have shown promise in preclinical and clinical settings, by enhancing patient survival rates. This review describes the evolving understanding of the cGAS-STING pathway’s involvement in tumor biology and therapy. Moreover, this review explores classical and non-classical STING agonists, providing insights into their mechanisms of action and potential for optimizing immunotherapy strategies. Despite challenges and complexities, the cGAS-STING pathway, a promising avenue for enhancing cancer treatment efficacy, has the potential to revolutionize patient outcomes.
keywords
Introduction
The human immune system plays critical roles in tumor development, progression, and metastasis, and is central to cancer therapy. Tumor immune surveillance relies predominantly on the innate immune system, a fundamental mechanism safeguarding the body against tumor intrusion by recognizing, regulating, and eliminating malignant cells. The efficacy of tumor antigen-specific T cells relies on signals originating from the innate immune system, which greatly influence T cells’ ability to combat tumor cells1.
In innate immunity, pattern recognition receptors (PRRs) detect molecules associated with infections, cellular stress, and tissue damage. Throughout tumor initiation and progression, cancer cells generate a plethora of damage-associated molecular patterns; these molecules interact with PRRs and subsequently trigger innate immune responses2. In innate immunity, the cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING) pathway plays an instrumental role. This pathway is a dedicated mechanism for sensing and signaling in response to specific DNA stimuli3. Substantial evidence indicates that cGAS detects double-stranded DNA (dsDNA) originating from deceased cancer cells, mitochondria, and other sources. This recognition event activates the STING signaling cascade and culminates in the production of type I interferon (IFN-α/β). IFN-α/β in turn orchestrate the maturation, differentiation, and mobilization of dendritic cells (DCs), macrophages, natural killer (NK) cells, effector T cells, and other immune effectors. Furthermore, the STING signaling pathway elicits the release of tumor antigens, thereby facilitating their cross-presentation by antigen-presenting cells4,5. Given its critical role in reshaping the tumor microenvironment (TME), STING has emerged as an attractive target for cancer immunotherapy.
Intervention strategies aimed at activating the cGAS-STING pathway, such as the use of pharmacological agonists, have achieved substantial advancements in both preclinical and clinical research settings. Activation of the cGAS-STING pathway has shown favorable tumor regression effects in clinical models, by evoking systemic anti-tumor responses, particularly when combined with immune checkpoint inhibitors (ICIs). These combination therapies have achieved remarkable improvements in survival rates in phase I clinical trials6,7. Consequently, STING pathway activation is considered a means of increasing tumor immunotherapy efficacy through mobilizing both innate and adaptive immune mechanisms.
In this review, we aim to comprehensively describe cutting-edge developments in exploiting the cGAS-STING pathway in tumor biology and therapy. We discuss the current research landscape of classical and non-classical STING agonists used for cGAS-STING pathway activation. These investigations have not only increased understanding of the mechanisms underpinning the cGAS-STING pathway in tumor immunity but also have provided a critical theoretical framework for developing more efficacious immunotherapy strategies. These efforts have the potential to revolutionize the efficacy of cancer treatments and enhance patient survival rates.
cGAS-STING signaling pathway
cGAS, a member of the nucleotidyl transferase family, contains unstructured N-terminal and C-terminal catalytic domains8. This enzyme is essential in the surveillance of diverse sources of dsDNA. After binding dsDNA, cGAS dimerizes, forming a cGAS-dsDNA complex comprising 2 cGAS molecules and 2 dsDNA molecules9. Crystallographic analysis has demonstrated that in the dsDNA-cGAS interaction, longer DNA strands have greater efficacy than shorter DNA strands in facilitating cGAS liquid-liquid phase separation and enzymatic activity, thus enabling cGAS to catalyze the synthesis of 2′3′-cGAMP from adenosine triphosphate (ATP) and guanosine triphosphate (GTP)10. Recent investigations have indicated that, although RNA does not activate cGAS in vitro, it potentiates cGAS phase separation. Particularly when dsDNA concentrations are limited, RNA fosters cGAS-containing phase separation and consequently increases cGAS activity11. In contrast to other closely related cyclic dinucleotides (CDNs), the product of cGAS, 2′3′-cGAMP, is as a distinctive dinucleotide featuring a phosphodiester bond between the 2′-hydroxyl of GMP and the 5′-phosphate of AMP, as well as another phosphodiester bond between the 3′-hydroxyl of AMP and the 5′-phosphate of GMP. These structural attributes enhance affinity for the human STING receptor8.
STING, encoded by the transmembrane protein 173 (TMEM173) gene, is a transmembrane protein with an N-terminal domain and a C-terminal domain, each bearing 4 transmembrane structures, and existing in a homodimeric configuration12. In its quiescent state, STING resides in the endoplasmic reticulum (ER) membrane and the outer mitochondrial membrane, and its C-terminus freely extends into the cytoplasm. After binding cGAMP, STING undergoes substantial conformational changes, which trigger its translocation from the ER to the ER-Golgi intermediate compartment, in a process crucial for signal transduction13. Subsequently, STING recruits TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3). TBK1 phosphorylates serine residue S366 in the C-terminus of STING, thereby activating IRF3 and prompting its translocation to the nucleus. IRF3 subsequently facilitates the transcription of IFN-α/β and immune-stimulating genes14. In addition to phosphorylation, palmitoylation of STING at C88/C91 is indispensable for assembling the signal complex, through facilitating STING aggregation in the Golgi apparatus15. Concurrently, STING activates IκB kinase (IKK) and consequently governs the production of NF-κB-mediated inflammatory factors16. A recent study has proposed that STING protein also functions as a hydrogen ion channel facilitating Golgi hydrogen ion efflux, thereby promoting non-classical autophagy and NLRP3 inflammasome activation17. Moreover, STING interacts with hexokinase 2 (HK2), a rate-limiting enzyme in glycolysis, and subsequently restrains the mitochondrial localization of HK2, thus inhibiting its activity and suppressing glycolysis (Figure 1)18.
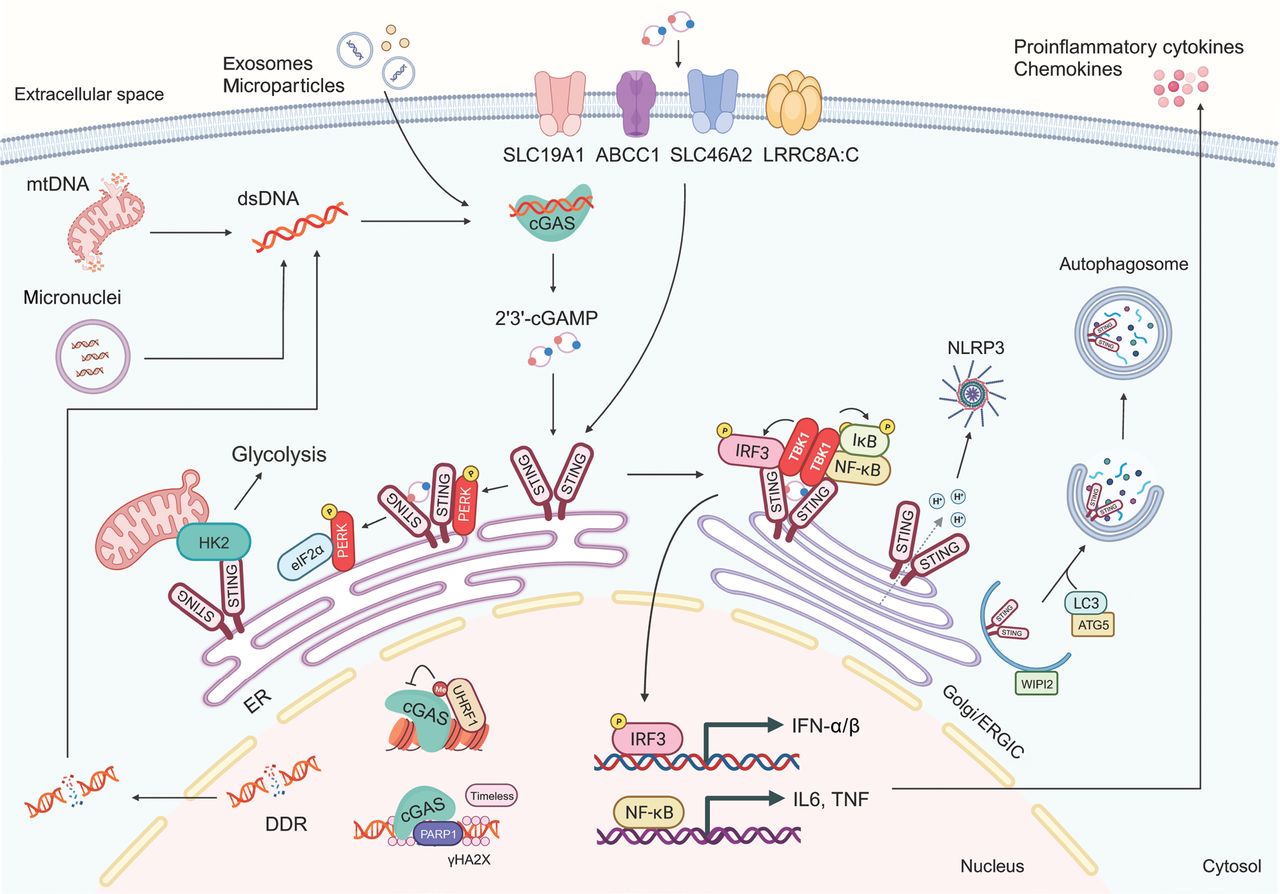
The cGAS-STING signaling pathway. Double-stranded DNA (dsDNA) derived from the nuclear DNA damage response (DDR), mitochondria, and micronuclear DNA activates cyclic GMP-AMP synthase (cGAS) and drives the synthesis of 2′,3′-cyclic GMP-AMP (cGAMP). After binding cGAMP, stimulator of interferon genes (STING) is translocated from endoplasmic reticulum (ER) to the Golgi and ER-Golgi intermediate compartment (ERGIC). Subsequently, STING recruits and activates TANK-binding kinase 1 (TBK1), which in turn phosphorylates interferon regulatory factor 3 (IRF3) and nuclear factor κB (NF-κB). IRF3 and NF-κB then translocate into the nucleus, where they facilitate the transcription of type I-interferon and immune-stimulating genes. In the ER, STING interacts with protein kinase R (PKR)-like ER kinase (PERK), thus leading to its activation. STING also functions as a hydrogen ion channel facilitating Golgi hydrogen ion efflux, thereby promoting non-classical autophagy and NLRP3 inflammasome activation. Moreover, nuclear cGAS inhibits DNA homologous recombination repair, whereas UHRF1 regulates the chromosomal untethering of cGAS. mtDNA, mitochondrial DNA; HK2, hexokinase 2; IL6, interleukin-6; TNF, tumor necrosis factor; ATG5, autophagy-related 5.
Additionally, in cGAS-STING signal transduction, the activity and stability of cGAS and STING are regulated by various post-translational modification mechanisms such as ubiquitination, phosphorylation, SUMOylation, and neddylation19–23. An in-depth exploration of the structural and biological facets of the cGAS-STING signaling pathway can be found in recent comprehensive reviews5,9,24.
The cGAS-STING pathway in cancer biology
Mechanisms of cGAS-STING pathway activation in cancer cells
A hallmark of cancer is the DNA damage response (DDR), which encompasses the recognition, signaling, and repair of DNA damage25. Emerging evidence has shown that endogenous DDR defects in cancer cells activate the cGAS-STING pathway. These mechanisms primarily involve the overproduction of abnormal DNA fragments in the nucleus, and the presence of micronuclei DNA or chromatin fragments associated with senescence in the cytoplasm26.
Extensive DNA damage, whether intrinsic or induced by treatments such as radiation or chemotherapy, produce dsDNA fragments that activate the cGAS-STING pathway27. Repair factors, including mutL homolog 1 (MLH1) and the excision repair cross-complementation group 1 (ERCC1), replication stress-associated SAM and HD domain-containing protein 1 (SAMHD1), and the ataxia-telangiectasia mutated (ATM)-checkpoint kinase, are closely associated with cGAS-STING activation28–30. Moreover, cancer cells tend to accumulate other types of abnormal DNA, such as extrachromosomal circular DNA elements and extrachromosomal telomere repeat DNA, which evoke robust IFN-α/β responses, and subsequently activate immune responses in macrophages and DCs31.
Mislocalized DNA, accompanied by chromatin fragments associated with senescence in the cytoplasm, is encapsulated within micronuclei in cells. In pre-cancerous or cancerous cells, structural membrane anomalies render micronuclei and senescence-associated cytoplasmic chromatin fragments susceptible to rupture and release of highly immunostimulatory dsDNA fragments that bind and activate cGAS32. Notably, particularly in the case of radiation therapy, DNA damage-induced micronucleus formation contributes to the expression of pro-inflammatory genes and cytokines, thereby leading to innate immune activation in mouse mammary carcinoma cells33. Diverse mechanisms have been identified in the autoactivation of cGAS that has encountered DNA in micronuclei in various cancer scenarios. For instance, mutations in the RecQ-like helicase BLM or defects in RNase H2 can lead to formation of micronuclei and cGAS-dependent IFN-α/β responses; consequently, patients with Bloom syndrome are susceptible to all types of cancer34. Liu et al.35 have revealed that DNA damage can lead to translocation of cGAS from the cytoplasm to the nucleus in a manner dependent on importin-α, whereas phosphorylation of cGAS at residue Y215 promotes its cytoplasmic retention. Inside the nucleus, cGAS is recruited to sites of dsDNA breaks, where it interacts with poly(ADP-ribose) and inhibits the formation of the poly-adenosine diphosphate ribose polymerase 1 (PARP1)-Timeless complex, thereby impeding homologous recombination. PARP inhibitors (PARPi) enhance cancer cell immunogenicity via the cGAS-STING signaling pathway in the context of ERCC1 or the breast cancer susceptibility genes (BRCA) defects. Several studies have indicated that changes in chromatin states induced by chemoradiotherapy significantly influence cGAS recruitment, whereas the inner nuclear membrane-anchored exonuclease three prime repair exonuclease 1 (TREX1) degrades DNA and mitigates the effects of IFN-α/β36,37. Recently, our group has shown that multiple cancer cells induce chromatin untethering and activity inhibition of cGAS through competitive uptake of methionine. Mechanically, cGAS undergoes methylation modifications at multiple sites, among which K362 methylation is dependent on methionine and S-adenosylmethionine. The chromatin binding protein ubiquitin-like, containing PHD and RING finger domains 1 (UHRF1) recruits methylated cGAS and promotes its chromosome binding, whereas short-term methionine restriction or targeted interventions with methyltransferase suppressor of variegation 3–9 homolog 1 (SUV39H1) decrease cGAS methylation, and consequently promote its chromosome dissociation and entry into the cytoplasm. Subsequently, cytoplasmic cGAS is again activated by dsDNA, thus enhancing the anti-tumor immune phenotype of radiation therapy and ICIs38.
Beyond the aforementioned abnormal DNA sources, other potential contributors may activate the cGAS-STING pathway in cancer cells. Tumors have been suggested to indirectly evoke cGAS-STING activation through endogenous retrotransposons, thereby leading to genome integrity impairment, accumulation of DNA damage products, or the formation of micronuclei39. For example, the upregulation of L1 retrotransposons is correlated with late-stage senescence and enhances cGAS-STING-dependent IFN-α/β responses—a process exacerbated by diminished TREX1 activity40. Additionally, cytosolic DNA released from perturbed mitochondria elicits cGAS-STING activity41.
In summary, abnormal DNA, although generally associated with promoting cell malignancy and tumor development, concurrently triggers innate immunity in cells in a cGAS-STING-dependent manner. Nevertheless, further investigation is necessary to gain a comprehensive understanding of the precise mechanisms governing these processes.
cGAS-STING facilitates crosstalk between cancer cells and the TME
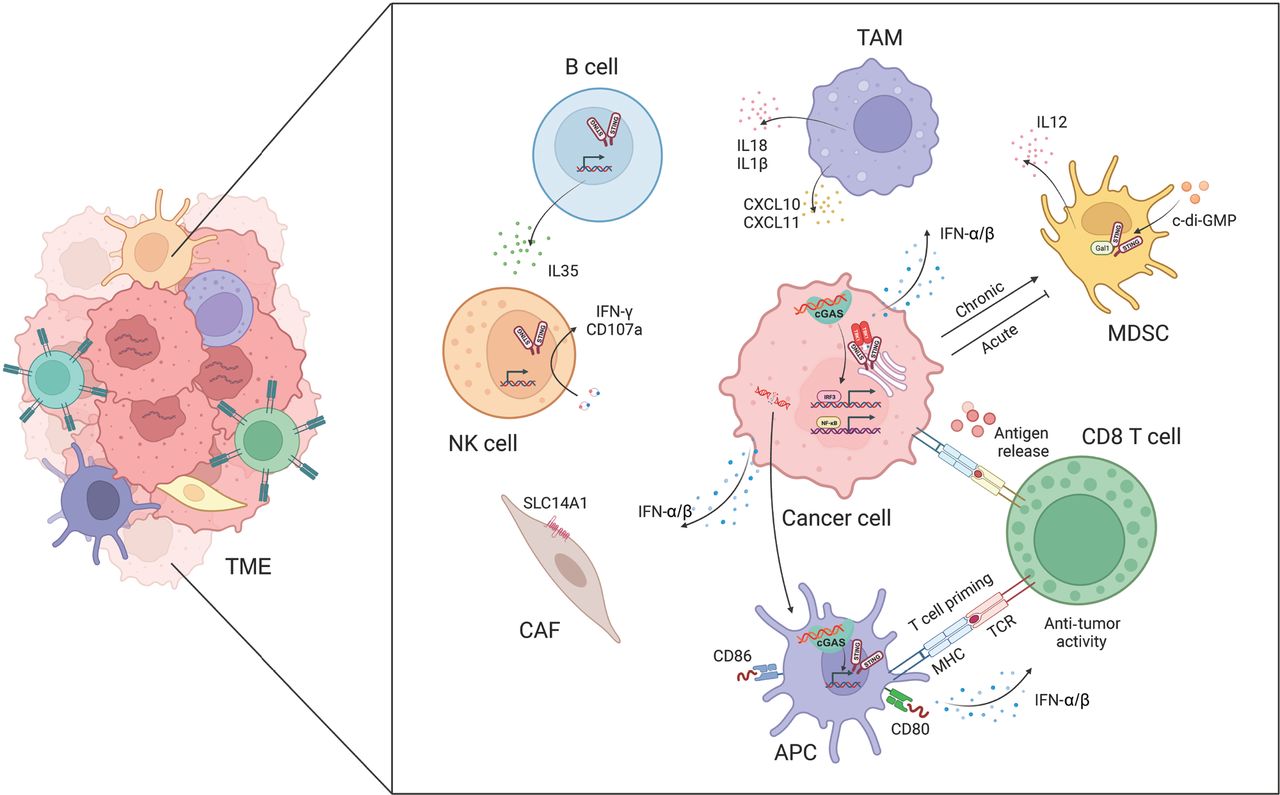
In cancer, the cGAS-STING pathway has multifaceted influences on tumor immunogenicity, and fosters interactions between cancer cells and various components of the TME. Among its critical anti-tumor effects, the cGAS-STING pathway elicits a potent IFN-α/β response, which is indispensable in activating immune cells in the TME, including DCs, T cells, and NK cells (Figure 2).
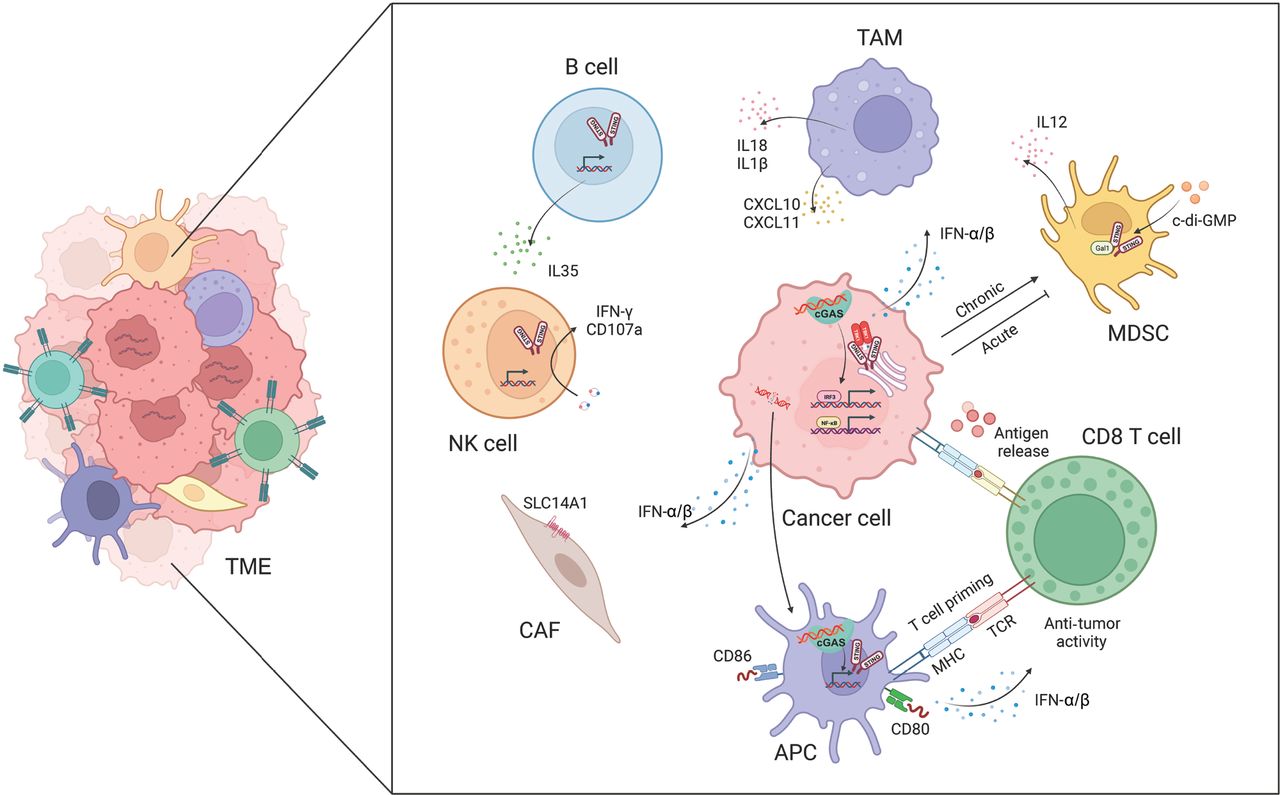
The cGAS-STING pathway facilitates crosstalk between cancer cells and the tumor microenvironment (TME). The activation of cGAS-STING promotes the release and presentation of cancer antigens, T cell migration and infiltration, and T cell recognition and cytotoxicity. The cGAS-STING pathway also regulates tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), natural killer (NK) cells, B cells, and cancer-associated fibroblasts (CAFs).
Activation of cGAS-STING promotes cancer antigen release and presentation
Under conditions of genomic instability, radiotherapy, or chemotherapy, dying cancer cells release cancer antigens, which trigger anti-tumor immune responses. DCs, as critical antigen-presenting cells in the immune system, are responsible for capturing, processing, and presenting antigens, and subsequently initiating T cell-mediated immune responses42. Tumor-derived DNA or cGAMP is internalized and processed by DCs that have infiltrated tumors; subsequently the cGAS-STING pathway is activated and the production of IFN-α/β is induced. This process drives DC differentiation and maturation, and concurrently enhances major histocompatibility complex class I (MHC-I) expression and facilitates DC-mediated cytotoxic T lymphocyte (CTL) anti-tumor responses43. Furthermore, the cGAS-STING pathway amplifies the expression of co-stimulatory molecules (e.g., CD40, CD80, and CD86) on DCs while simultaneously decreasing the expression of inhibitory molecules, such as programmed death-ligand 1 (PD-L1)44. Mn2+ has been found to augment the maturation of both DCs and macrophages, facilitate the presentation of tumor-specific antigens, promote the differentiation and activation of CD8+ T cells and NK cells, and intensify the presence of memory CD8+ T lymphocytes45. In ultraviolet (UV)-irradiated ovalbumin (OVA)-expressing EG7 thymoma cells, IFN-α/β sustains the intracellular presence of antigens in CD8α+ DCs. These DCs engulf apoptotic EG7 cells, thereby enhancing the cross-presentation of OVA antigens. The deletion of mitochondrial transcription factor A in DCs leads to mitochondrial dysfunction and leakage of mitochondrial DNA (mtDNA) into the cytoplasm, and subsequently activates the cGAS-STING pathway in DCs. This enhanced antigen presentation effectively reverses the immunosuppressive TME46.
Activation of cGAS-STING promotes T cell migration and infiltration
In the immune system’s recognition and elimination of cancer cells, T cells must initially mobilize and infiltrate tumor tissue. Under the induction of STING signaling, various chemokines [e.g., C-X-C motif chemokine ligand 9 (CXCL9), CXCL10, and C-C motif chemokine ligand 5 (CCL5)] play critical roles in stimulating the migration and infiltration of CTLs47. Administration of STING agonists induces the generation of IFN-α/β signals in DCs, thereby stimulating the secretion of chemokines such as CXCL9 and CXCL10, which further promote CTL migration into tumor tissue48. Immunotherapy using c-di-GMP activates IFN-α/β signal transduction in CD11b+ cells and facilitates the infiltration of CD8+ T cells into glioblastoma49. In mouse models, treatment with cGAMP results in the accumulation of a CD45+CD11bmidLy6C+ macrophage subpopulations in the TME. These macrophages express numerous chemokines that recruit T cells, thereby modulating the infiltration of CD8+ T cells and promoting anti-tumor immunity50. Moreover, Bruand et al.51 have revealed that the elimination of endogenous STING in tumors decreases neoangiogenesis, augments CD8+ T cell infiltration, and reverses resistance to dual immune checkpoint blockade therapy. Ariadne RBR E3 ubiquitin protein ligase 1 (Arih1) mediates the ubiquitination and degradation of DNA PKcs, and consequently triggers STING pathway activation, promotes the infiltration of cytotoxic T cells, inhibits tumor growth, and renders tumors sensitive to PD-L1 blockade52. The nuclear receptor Rev-erb alpha (NR1D1) fosters the accumulation of cytoplasmic DNA fragments induced by DNA damage, and subsequently activates the cGAS-STING signaling pathway. This enhancement results in increased production of IFN-α/β, and expression of chemokines such as CCL5 and CXCL10, which in turn regulate the infiltration of CD8+ T cells and NK cells53. These findings further underscore the crucial role of the cGAS-STING signaling pathway in promoting T cell migration and infiltration.
Enhancing T cell recognition and cancer cell killing through cGAS-STING activation
T cells play essential roles in the immune response against cancer cells, primarily by recognizing tumor-specific antigens presented on MHC-I molecules via their T cell receptors. Multiple studies have demonstrated the importance of STING pathway activation in increasing CTL engagement and eliciting robust anti-tumor responses.
In murine models, treatment of CT26 colorectal cancer or 4T1 breast cancer cells with radiation has been demonstrated to initiate effective STING-dependent CTL responses43. Platinum-based chemotherapy has similarly been shown to activate STING and increase expression of MHC-I in cancer cells. This heightened MHC-I expression facilitates the recognition of cancer cells by CTLs, thereby amplifying CTL-mediated anti-tumor effects. In contrast, ionizing radiation upregulates the expression of PD-L1 in liver cancer cells via activation of the STING-TBK1-IRF3 immune pathway. This upregulation of PD-L1 dampens CTL activity and ultimately shields cancer cells from immune-mediated clearance. Furthermore, cGAMP treatment has been found to augment the anti-tumor effects of radiation in a hepatocellular carcinoma mouse model, thus bolstering the cytotoxic capabilities of tumor-specific CD8+ T cells54. Huang et al.55 have recently developed a collaborative nanoplatform promoting the infiltration of CD8+ T cells into tumors. This platform restores T cell function by modulating tyrosine metabolism and activating the STING pathway. Moreover, targeting STING for systemic therapy in mice eliminates tumor cell dormancy and metastasis in a T cell and NK cell dependent manner56. Interestingly, intrinsic STING in T cells also decreases apoptosis and enhance long-term T cell protective immunity57. Collectively, these findings highlight the promise of activating the STING pathway as a potential strategy for inducing potent anti-tumor immune responses.
Regulation of tumor-associated macrophages (TAMs) by cGAS-STING activation
TAMs play a major role in tumorigenesis, and targeting TAM function is a potential therapeutic approach to mitigating tumor metastasis58. Notably, studies have illuminated the influence of cGAS-STING activation on TAM behavior. Ohkuri et al.50 have reported that injection of cGAMP into mouse tumor tissue results in macrophage aggregation, whereas this effect is absent in STING knockout mice. Notably, the anti-tumor effects induced by cGAMP are abrogated after macrophage depletion in mice. Moreover, ATP enhances the transport of extracellular cGAMP to macrophages through the ATP-gated P2X purinoceptor 7 (P2X7) receptor (P2X7R) and subsequently leads to STING activation59. Further analysis has revealed that STING-induced tumor-associated migratory macrophages exhibit elevated levels of T cell-recruiting chemokines, such as CXCL10 and CXCL11, which facilitate CD8+ T cell recruitment to tumor sites. STING activation has been shown to reprogram TAMs into an M1 phenotype, and to transform immunologically “cold” peritoneal tumors into T cell-inflamed tumors in an IFN-α/β-dependent manner60. In lung and breast cancers, the STING pathway has been found to be deactivated and positively correlated with macrophage markers. Treatment with vanillic acid has been demonstrated to stimulate the STING/TBK1/IRF3 pathway, thus inducing IFN-α/β production and promoting macrophage polarization toward an M1 phenotype, and enhancing phagocytosis and apoptosis induction61. In a model of liver cancer metastasis, STING activation has been found to influence TAM polarization and to promote the nuclear translocation of transcription factor EB via interferon-associated GTPase 1. This process affects macrophages’ ability to suppress tumor metastasis62. Moreover, STING activation assists macrophages in secreting interleukin-18 (IL-18) and interleukin-1β (IL-1β), thereby optimizing NK cell anti-tumor activity through promoting the expression of 4-1BBL in macrophages and 4-1BB in NK cells63. Additionally, the disruption of protein phosphatase 2A catalytic subunit (PP2Ac) in glioblastoma cells enhances dsDNA production and cGAS-IFN-α/β signaling. This process increases MHC-I expression, decreases immunosuppressive TAMs, and sensitizes tumors to immune checkpoint blockade and radiotherapy64. Ho et al.65 have found that, in TAMs, protein PP2A and its specific B regulatory subunit Striatin 4 (STRN4) negatively regulate STING-dependent IFN-α/β production. The diminished accumulation of immunosuppressive macrophages and increased IFN-α/β production in mice lacking macrophage PP2A ultimately decrease tumor progression. Moreover, STING activation has been found to involve the Hippo kinase mammalian sterile 20-like kinase 1/2 (MST1/2), on the basis of observations that STING agonists induce the dissociation of PP2A from MST1/2 in normal macrophages but not in TAMs. In breast cancer cells with BRCA1 mutations, TAMs undergo M2 polarization, which suppresses cancer cell DNA damage induced by PARPi. This suppression decreases dsDNA fragment production and synthetic lethality, and weakens STING-dependent anti-tumor immunity66. These findings illuminate the critical role of cGAS-STING-regulated macrophage activity in anti-tumor immunity.
Activating cGAS-STING to regulate the activity of other immune cells
NK cells have emerged as important components in cancer immunotherapy. Early investigations underscored the importance of STING expression in non-tumor cells for NK cells′ ability to selectively target and eliminate cancer cells rather than normal cells67. Across various tumor models, STING agonists have shown remarkable potential in enhancing NK cell-mediated anti-tumor responses in a manner independent of CD8+ T cells68. These agonists not only promote NK cell migration and cytotoxicity, but also augment the activation of NK cells in combination with CAR-NK cells69. Furthermore, they potentiate trastuzumab-mediated NK cell activation and DC maturation via STING activation70, and directly activate NK cells, thereby increasing the sensitivity of pancreatic cancer cells to NK cell-mediated cytotoxicity71. Intriguingly, cGAMP induces B cell expression of IL-35 in an IRF3-dependent but IFN-α/β-independent manner. IL-35 secreted by B cells inhibits tumor growth but concurrently suppresses NK cell proliferation, thus tempering NK-driven anti-tumor responses72. Recently, Lu et al.73 have reported that cancer cell derived mtDNA recognition by cGAS triggers the NK cell-intrinsic STING pathway, and consequently promotes antitumor responses and maintains a reservoir of TCF-1+ NK cells.
Myeloid-derived suppressor cells (MDSCs) are a highly heterogeneous population of immature immune cells that support tumor growth and progression by impeding the proliferation and function of effector immune cells. Notably, the cGAS-STING signaling axis influences the recruitment, expansion, and function of MDSCs74. c-di-GMP can be used to target MDSCs: even low doses of this compound significantly increase the production of IL-12 by MDSCs75. Moreover, STING activation in cancer cells and MDSCs enhances the expression of suppressor of cytokine signaling 1 (SOCS1), which restrains the induction of MDSCs derived from nasopharyngeal carcinoma76. The STING pathway, after activation by radiation exposure, leads to the infiltration of MDSCs and, paradoxically, immune suppression, thus ultimately contributing to tumor radioresistance77. Additionally, Galectin-1 (Gal1) plays roles in stabilizing STING protein, sustaining NF-κB activation in cancer cells, perpetuating the accumulation of MDSCs in the pre-metastatic niche, and eliciting CXCL2-mediated migration of MDSCs78.
Cancer-associated fibroblasts (CAFs) are instrumental in shaping responses to cancer treatment and influencing patient outcomes. Activation of the cGAS-STING pathway in tumors plays a major role in defining the characteristics of CAFs, thus resulting in marked upregulation of CAF markers that confer tumor-suppressive properties79. Activation of the cGAS-STING pathway further induces the production of interferons and consequently drives overexpression of the urea transporter protein human solute carrier family 14 member 1 (SLC14A1) in a specific subset of CAFs. Notably, targeting STING hinders the formation of SLC14A1-positive CAFs and renders tumor cells more susceptible to chemotherapy80. Recently, Dora et al.81 have reported CAFs expressing STING in the stroma, and suggested that many STING-expressing cells in the stroma might be CAFs in small cell lung cancer tissues. Moreover, STING and IRF3 in stromal fibroblasts can sense genomic stress in cancer cells82, and targeting IRF3 in CAFs has been found to restore oncolytic herpes simplex virus function82.
Strategies for harnessing the cGAS-STING pathway in cancer therapy
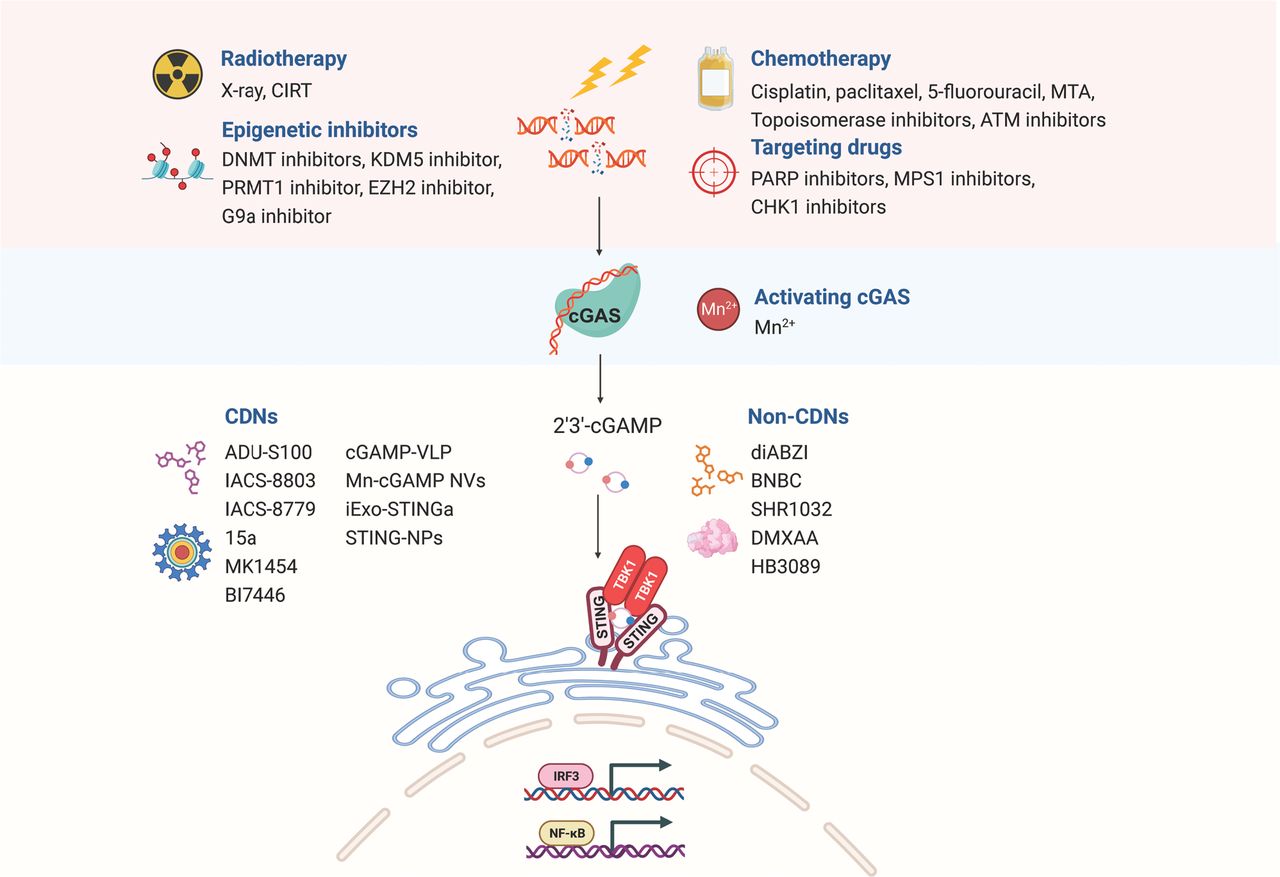
As described above, the presence of extracellular DNA from various sources, including DNA damage, genetic instability, mitochondria, and exosomes, renders cancer cells susceptible to cGAS-STING pathway activation. cGAS-STING pathway can also be triggered by exogenous stressors, such as radiation and chemotherapy, which induce the accumulation of micronuclei and cytoplasmic DNA fragments26. Preclinical investigations have demonstrated that stimulating the cGAS-STING pathway with cytotoxic agents or molecular inhibitors has promise in improving cancer treatment outcomes, particularly in combination therapies with ICIs, thus offering a potential breakthrough in cancer therapy (Figure 3).
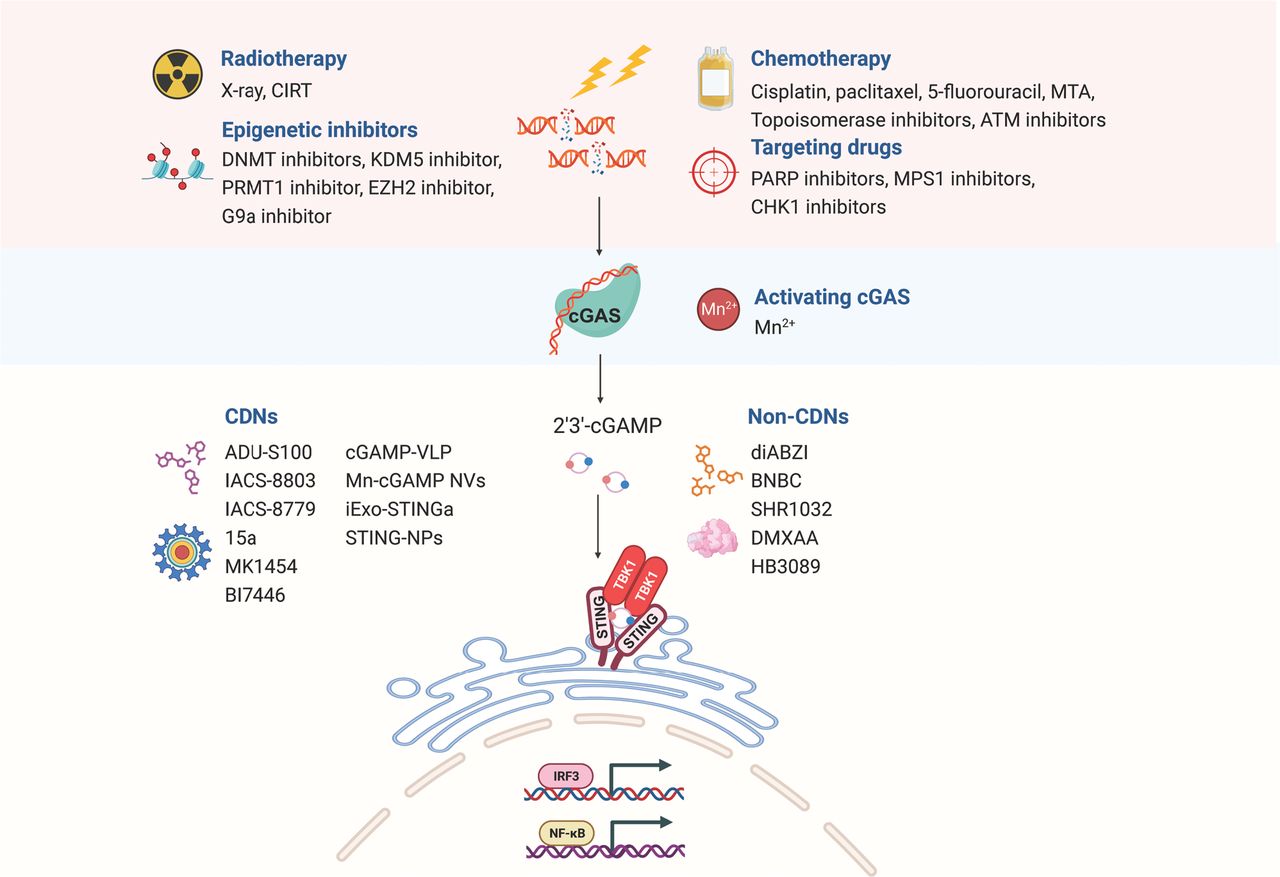
Schematic diagram of harnessing the cGAS-STING pathway in cancer therapy. The accumulation of cytoplasmic and micronuclear DNA fragments induced by radiotherapy, chemotherapy, targeted therapy, and epigenetic therapy activates the cGAS-STING pathway, thus exerting anti-tumor activity. STING agonists independent of cGAS can also be used to directly activate the STING pathway; these agonists include natural cyclic dinucleotide (CDN) and non-CDN agonists. CIRT, carbon ion irradiation; DNMT, DNA methyltransferase; KDM5, lysine-specific demethylase 5; PRMT1, protein arginine methyltransferase 1; EZH2, enhancer of Zeste 2 Polycomb repressive complex 2 subunit; MTA, microtubule-targeting agent; PARP, poly-adenosine diphosphate ribose polymerase; MPS1, monopolar spindle 1; ATM, ataxia telangiectasia-mutated gene; CHK1, checkpoint kinase 1.
Strategies based on dsDNA-dependent cGAS-STING pathway activation
Radiotherapy-induced cGAS-STING activation
Radiotherapy, by inducing apoptosis and DNA damage in cancer cells, disrupts DNA structure. Furthermore, radiotherapy evokes extensive DNA damage resulting in cell death and the release of DNA fragments. Consequently, cGAS-STING pathway activation culminates in the initiation of an immune response83. Notably, IFN-α/β has a critical role in reshaping the TME after radiotherapy. As radiation doses escalate, the expression of dsDNA, 2′3′-cGAMP, cGAS, and immune-stimulating genes concomitantly increases in the cytoplasm, alongside elevated levels of IFN-β in the cellular supernatant84. Mitochondria are a recognized source of reactive oxygen species, and radiation-induced oxidative stress can lead to the leakage of mtDNA into the cytoplasm and activation of the cGAS-STING pathway85. However, that mtDNA is notably not the primary target of radiation, and the probability of DNA double-strand breaks in mtDNA is approximately 0.03% under conditions of 1 Gy of gamma radiation or dense ionizing radiation86. After dsDNA breakage, micronuclei form during mitosis as a consequence of cell cycle progression, preceding the activation of inflammatory signals87. Therefore, accumulation and sensing of micronuclei DNA are crucial triggers of the cGAS-STING pathway, which ultimately culminates in IFN-α/β signaling32. In summary, radiation-induced micronuclei and dsDNA are indispensable for the sensing of cGAS and activation of STING, thereby facilitating the anti-tumor immune response.
The DNA exonuclease TREX1 is a crucial upstream regulator of radiation-induced cGAS-STING-mediated anti-tumor immunity. Radiation doses exceeding 12–18 Gy induce the expression of TREX1 in various cancer cells, and mitigate immunogenicity by degrading dsDNA accumulated in the cytoplasm during radiation exposure88,89. The classical NF-κB signaling pathway has a central role in driving radiation-induced cGAS-STING pathways. Evaluation of tumor growth after irradiation has revealed that the absence of intact classical NF-κB pathways does not have anti-tumor effects. In contrast, intact NF-κB pathways hinder tumor growth, thus underscoring the role of atypical NF-κB pathways in promoting radiation-induced anti-tumor immunity90. In a primary mouse model of soft tissue sarcoma, depletion of alpha-thalassemia mental retardation X-linked (Atrx), a chromatin remodeling protein and tumor suppressor, significantly intensifies radiation-induced persistent DNA damage, telomere dysfunction, and mitotic catastrophe. Remarkably, this process coincides with a pronounced impairment in the cGAS-STING-interferon signaling pathway91.
Effects of chemotherapy on cGAS-STING pathway activation
Chemotherapy uses various agents, such as cisplatin, paclitaxel, and 5-fluorouracil, to disrupt DNA replication or induce double-strand break repair processes, thus resulting in cGAS-STING pathway activation. This activation stems from the cytoplasmic accumulation of DNA fragments, and leads to chemokine secretion, antigen presentation, and initiation of IFN-α/β signaling21. For example, cisplatin and gemcitabine induce the STING-dependent interferon pathway in nasopharyngeal carcinoma, and subsequently enhance MHC-I expression and IL-1β production92. In bladder cancer, cisplatin-induced cGAS-STING signaling not only inhibits cancer cell proliferation, but also promotes CD8+ T cell and DC infiltration93. Microtubule-targeting agents such as paclitaxel induce pro-inflammatory responses via cGAS-STING, activate interferon signaling, and trigger downstream immune responses94. Topoisomerase inhibitors, including etoposide and amsacrine, activate the cGAS-STING pathway by disrupting DNA replication and inducing double-strand breaks. Etoposide induces NF-κB and STING-dependent IFN-α/β signaling, thereby enhancing anti-tumor T cell responses95. However, amsacrine-induced DNA damage in a hypoxic environment exhibits differential effects in estrogen/progesterone receptor-positive and triple-negative breast cancer cells. These findings highlight the complex interplay of chemotherapy with the cGAS-STING pathway96. Additionally, ATM kinase, a regulator of DNA damage responses, plays a major role in cGAS-STING activation, and ATM inhibitors have shown potential in enhancing cancer cell sensitivity to radiation and promoting interferon responses via the cGAS-STING/TBK1 pathway30,97.
In summary, these findings collectively underscore the critical role of the cGAS-STING pathway in the context of radiation and chemotherapy for cancer treatment.
Exploiting targeted therapies to activate the cGAS-STING pathway for antitumor responses
Loss of BRCA1 leads to transcriptional reprogramming in cancer cells, and intrinsic inflammation involving STING and IFN-α/β51. PARP1 mediates DNA repair through homologous recombination, and PARPi enhance genomic instability, thus leading to specific autologous DNA cytoplasmic leakage in cancer cells with homologous recombination defects associated with BRCA1/2 mutations; subsequently, immune infiltration increases, and tumor clearance is promoted98. PARPi modulate the TME by activating the cGAS-STING pathway and altering the balance of immune-stimulating signals. Low-dose CAR-T cell therapy therefore induces effective tumor regression99, and blocking immune checkpoints further enhances the effect of PARPi100. Clinically, the PARPi olaparib and rucaparib exhibit properties of cell-autonomous immune modulation in ERCC1-deficient non-small cell lung cancer and BRCA1-deficient TNBC cells: they generate cytoplasmic chromatin fragments with micronuclear characteristics, and activate cGAS/STING, downstream IFN-α/β signaling, and CCL5 secretion101. Recent research has reported that the high-affinity PARP inhibitor thioparib targets multiple PARPs, including PARP1, PARP2, and PARP7, and displays high anti-tumor activity in vitro and in vivo in cells that bear homologous recombination defects, and are either sensitive or resistant to PARPi. Thioparib-induced STING/TBK1-dependent signal transducer and activator of transcription 1 (STAT1) phosphorylation targets PARP7, induces a robust IFN-α/β response, and slows tumor growth in immunocompetent mouse models102. Olaparib in combination with the WEE1 inhibitor adavosertib triggers anti-tumor immune responses, including the STING pathway. When used in conjunction with STING agonists, this therapy further enhances persistent tumor regression in BRCA1/2 wild-type TNBC mouse tumor models and significantly improves survival outcomes103.
Monopolar spindle 1 (MPS1) is a critical component of the spindle assembly checkpoint. In the presence of MPS1 inhibitors, cancer cells continue to divide, thus resulting in abnormal spindle formation and the appearance of numerous chromatin bridges and micronuclei, which are potent activators of the cGAS/STING pathway104. Kitajima et al.105 have found that KRAS-LKB1 mutant lung cancer cells minimize the intracellular accumulation of 2′3′-cGAMP; consequently, downstream STING and STAT1 activation is avoided. However, inhibiting MPS1 effectively re-engages the STING pathway through micronucleus generation, thus effectively restoring T cell infiltration, enhancing anti-PD-1 efficacy, and producing lasting responses.
The checkpoint kinase 1 (CHK1) inhibitor prexasertib induces DNA damage in cancer cells and activates the STING/TBK1/IRF3 innate immune pathway106, thereby increasing levels of chemokines such as CXCL10 and CCL5, and inducing activation of cytotoxic T lymphocytes107. Another study has reported that CHK1 inhibition increases TBK1 phosphorylation but not IRF3 phosphorylation, and does not induce IRF or NF-κB reporter gene activation108. In a phase II trial on recurrent ovarian cancer, treatment with prexasertib has been found to upregulate activation markers of the STING pathway such as TBK1, which correlates with prolonged progression-free survival (9 months vs. 3 months, P = 0.003)109.
In summary, these studies have suggested that PARP, MPS1, and CHK1 inhibitors promote anti-tumor effects by activating the cGAS-STING pathway.
Harnessing epigenetic modifiers to activate the cGAS-STING signaling pathway
Epigenetic modifications such as DNA methylation and histone modifications regulate gene expression states and cellular functions. Because of the frequent epigenetic silencing of cGAS and STING genes in their promoters, alleviating this inhibition makes cells more sensitive to synthetic STING agonists and DNA-damaging agents110. A close correlation has been observed between cGAS-STING signaling and methylation changes, particularly those involving histone lysine demethylase 5 (KDM5)111. Inhibitors of KDM5 activate STING expression in mouse colorectal cancer cells and inhibit tumor growth in a STING-dependent manner112. Falahat et al.113, through whole-genome DNA methylation analysis, have discovered that hypermethylation of the promoter regions of cGAS and STING genes mediates their co-transcriptional silencing, thereby leading to widespread impairment of STING signaling in melanoma cells. DNA methyltransferase inhibitors reverse this inhibition and restore STING activity. Furthermore, in STING-defective melanoma cells, demethylation-mediated restoration of STING signaling enhances antigenicity by upregulating MHC class I molecules, and consequently mediates resistance to TIL-based immunotherapies113. In endometrial cancer, histone deacetylase HDAC3 interacts with β-estradiol-ERα and induces histone 3 lysine 4 deacetylation at the STING promoter, thus decreasing STING expression. Inhibiting HDAC3 increases STING expression and suppresses tumorigenesis114. A recent study has reported that protein arginine methyltransferase PRMT1 methylates cGAS at a conserved R133 residue, prevents cGAS dimerization, and inhibits cGAS/STING signaling in cancer cells. Inhibitors of PRMT1 activate cGAS/STING-dependent DNA sensing signals, and enhance the transcription of type I and type II interferon response genes115. In TP53-mutated acute myeloid leukemia (AML), the combined effects of histone modification and polyploidy trigger cGAS-STING pathway activation, lead to the secretion of cytokines and chemokines, and prompt the activation of T cells and macrophages under coculture with AML cells116.
In addition to reactivating cGAS-STING, epigenetic modifications activate dsDNA and dsRNA sensing pathways. For instance, m6A methylation affects the secondary structure of DNA and alters the immunogenicity of cytoplasmic DNA. The response to m6A-methylated DNA is dependent on the cGAS-STING signaling axis, but is independent of MyD88/TRIF and interferon-beta promoter stimulator-1 (IPS-1) signaling117. Single or combined treatment with the small molecule inhibitors UNC1999 (EZH2 inhibitor) and UNC0642 (G9a inhibitor) leads to dual inhibition of H3K9me2 and H3K27me3, thus suppressing the formation of cytoplasmic chromatin fragments and consequently inhibiting the cGAS-STING pathway118. Chen et al.119 have reported that glucose, a co-factor binding methyltransferase NSUN2, promotes NSUN2 oligomerization and activation, and maintains overall m5C RNA methylation, including TREX2. This stabilization of TREX2 limits cytoplasmic dsDNA accumulation and cGAS/STING activation, and consequently promotes tumorigenesis and resistance to anti-PD-L1 immunotherapy. Therefore, similarly to DNA-damaging agents, epigenetic inhibitor therapy may transition cancer cells from an immunosuppressive state to an active state while concurrently activating the cGAS-STING signaling pathway.
Therapeutic strategies for STING pathway activation independent of cGAS
The activation of STING does not rely solely on the presence of cGAS. Most STING agonists have been shown to directly activate STING signaling independently of cGAS. These agonists are synthesized as prototypes of natural ligands called CDNs, similar to the binding of cGAMP to STING. However, natural CDN agonists have drawbacks limiting their application, including poor cell targeting, low stability, and inefficient transport. Additionally, genetic variations in STING alleles affect the sensing and response to exogenous and endogenous CDNs120. Therefore, interest is growing in CDN-based STING synthetic agonists, to enhance their stability and therapeutic efficacy.
Unleashing the power of CDN agonists in antitumor immunity
CDNs are natural agonists of the STING pathway that activate the cGAS-STING pathway in the innate immune system. Various CDN molecules, such as cGAMP, c-diGMP, and c-diAMP, play crucial roles in regulating immune responses and suppressing tumor proliferation. cGAMP activates the cGAS-STING-IRF3 pathway in animal models, reshapes the tumor immune environment, and prevents tumor metastasis by reversing epithelial-mesenchymal transition and inhibiting the PI3K/AKT pathway121. Administration of cGAMP triggers the activation of STING and the production of IFN-β in bone marrow cells and B cells67. cGAMP also enhances the anti-tumor activity of 5-FU while significantly decreasing its toxicity122. Several studies have indicated that depleting extracellular cGAMP decreases the infiltration of tumor-associated immune cells and eliminates the effectiveness of radiation therapy. The extracellular hydrolase ENPP1 has a role in clearing cGAMP, and the use of ENPP1 inhibitors enhances the synergistic effect of extracellular cGAMP with radiation, thus delaying tumor growth123,124.
c-di-GMP targets MDSCs and cancer cells. Low-dose c-di-GMP increases MDSC production of IL12, whereas high doses of c-di-GMP (0.3-3 mmol/L) directly kill cancer cells, partly by activating thioredoxin-375. c-di-GMP also enhances the immunogenicity and anti-tumor effects of peptide vaccines against murine B16 melanoma125. Another CDN, c-di-AMP, binds STING and activates downstream IFN pathways in STING-positive metastatic breast cancer cells, thus inducing the translocation of IRF-3 to mitochondria, initiating caspase-9-mediated cell death, and inhibiting clonogenicity of TNBC cells126. Additionally, c-di-AMP induces high levels of antigen-specific IgG antibodies and elicits potent CTL, Th1, and IFN-γ-producing CD8+ memory T cell responses127.
To increase the stability and delivery efficiency of CDNs, researchers have designed various carriers. Intratumoral injection of virus-like particles containing cGAMP (cGAMP-VLPs) induces the differentiation of circulating tumor-specific T cells and decreases regulatory T cells (Tregs), thereby resulting in complete and sustained tumor eradication in the absence of Treg depletion128. Thiolated and Mn2+-coordinated cGAMP nanovaccines (Mn-cGAMP NVs) enable direct cytoplasmic co-delivery of cGAMP and Mn2+, and enhance the anti-tumor immune response129. Compared with standalone STING agonists, an exosome-delivered STING agonist, the cyclic GMP-AMP (iExoSTINGa) system, exhibits superior uptake by DCs, and leads to the accumulation of activated CD8+ T cells and enhanced anti-tumor immune responses130. STING activation nanoparticles (STING-NPs) developed by Shae et al.131 have been found to enhance cytoplasmic delivery of the endogenous CDN ligand cGAMP, and to amplify STING signaling in the TME and sentinel lymph nodes. Moreover, STING-NPs have been found to increase the half-life of cGAMP by 40-fold, thus facilitating accumulation in tumors. In a B16-F10 melanoma model, STING-NPs have been found to increase the response rate to PD-L1 antibodies and to significantly improve median survival132. Dosta et al.133 have recently designed poly(β-amino ester)-based nanoparticles (NPs) covalently associated with CDNs, which enable the release of STING agonists after reaching target immune cells. When absorbed by target immune cells in the TME and secondary lymphoid organs, CDN-NPs confer long-term immunity against future tumors. Moreover, the KL-7 peptide, derived from Aβ amyloid precursor fiber, self-assembles into nanotubes (PNTs) for loading and delivering c-di-GMP; the effectiveness of this therapy is significantly enhanced by stimulating the secretion of IL-6 and INF-β, along with phosphorylation of STING (S365) protein, upregulating CD4 and CD8 cytotoxic T cell killing of tumors, and enhancing the immune response in tumor tissues134. Additionally, DC-targeting STING agonist delivery systems have been designed. For example, the PLGA/STING@EPBM nanovaccine significantly enhances antigen cross-presentation of Clec9a+ DCs135, and a CD103+ DC targeted CDN formulation generates strong immune stimulation, even in relatively “cold” tumors during systemic administration136.
In comparison to natural CDNs, CDN-based synthetic STING agonists have enhanced stability and therapeutic efficacy. ADU-S100, a modified CDN substance that has entered clinical trials, activates the STING pathway in vitro and in vivo, and displays good stability and lipophilicity in various animal models. When injected into tumors, ADU-S100 activates potent CD8+ T cell responses, and induces the production of inflammatory cytokines and chemokines137,138. In a phase I clinical trial (NCT02675439), ADU-S100 has demonstrated good tolerance in patients with late-stage solid tumors and lymphoma, but the overall response rate for single tumor injections was lower than expected. Furthermore, whether the concentration of ADU-S100 delivered locally was equivalent to the concentration required for activity in preclinical models is unclear139. Additionally, carriers for systemic delivery of ADU-S100, such as DOTAP/cholesterol liposomes140, functionalized porphyrin-based nanoparticles (NP-AS)141, and biodegradable ADU-S100-loaded implants142, have shown promising preclinical efficacy.
Beyond ADU-S100, Ager et al.143 have reported 2 highly efficient STING agonists, IACS-8803 and IACS-8779, which have demonstrated potent activation of the STING pathway in vitro and superior systemic anti-tumor responses in a melanoma mouse model. STING agonist 15a, a synthetic CDN structurally distinct from natural ligands, with optimized properties for intravenous administration, has potent anti-tumor efficacy through cytokine secretion and activation of CD8+ cytotoxic T cells′ adaptive immune response144. MK-1454, a CDN synthesized by using bacterial cell lysates overexpressing cyclic GMP-AMP synthase145, shows potent upregulation of tumor cell cytokines and effective anti-tumor activity146. BI 7446 (CDN 13) activates all 5 STING variants in cell assays and induces long-term, immune-mediated tumor rejection in mice147.
In conclusion, CDN agonists, as key factors in STING pathway activation, have notable potential in anti-tumor therapy. With continued optimization of their performance and delivery modes, CDNs are expected to play greater roles in clinical applications.
Beyond CDNs: exploring novel STING agonists for cancer therapies
Non-CDN STING agonists are also gaining attention in cancer research. Ramanjulu et al.148 have identified the dimeric aminobenzimidazole diABZI, which, through high-throughput screening, has been found to enhance STING binding. DiABZI has robust anti-tumor effects and elicits complete tumor regression in mouse models, particularly when administered intravenously, thus providing a treatment option for more patients with cancer. Compared with ADU-S100, a novel small molecule agonist, SHR1032 displays high activity in human cells of different STING genotypes, and induces IFN-β production and strong anti-tumor effects149. The small molecule compound 6-bromo-N-(naphthalen-1-yl)benzo[d][1,3]dioxole-5-carboxamide induces proinflammatory cytokine responses in a manner dependent on STING, and consequently regulates the activation of CD4+ and CD8+ lymphocytes150. The multivalent STING agonist PC7A bind a non-competitive STING surface site different from the cGAMP binding pocket, and subsequently induces tumor responses dependent on STING expression and CD8+ T cell activity151. The flavonoid-based compound vadimezan (DMXAA) interacts with STING and produces anti-tumor effects in various mouse models152. For instance, a single intratumoral injection of DMXAA results in sustained cure in as many as 60% of mice carrying undifferentiated pleomorphic sarcoma; moreover, immune phenotyping has indicated enriched lymphocyte responses in tumors at multiple time points after treatment153. Despite significant results in preclinical and phase I/II clinical trials, DMXAA failed in phase III clinical trials, because of its strong affinity for mSTING and lack of affinity for hSTING152. Other potential STING agonists include HB3089154, ganciclovir155, and a series of 1H-pyrrole-3-carbonitrile derivatives156.
In summary, non-CDN STING agonists are gaining attention in cancer research. They offer new opportunities for anti-tumor therapies, particularly for patients who may not respond to CDN-based agonists. As research in this area continues, these novel STING agonists hold promise in the development of more effective cancer treatments.
Combination treatments with cGAS-STING pathway targeting and ICIs
The synergistic approach of targeting the cGAS-STING pathway in combination with ICI treatment has intriguing possibilities in cancer therapy. Notably, patients treated with paclitaxel and ICIs demonstrate a significant correlation between elevated baseline cGAS expression and positive treatment responses157. Cisplatin, via a DNA damage-mediated cGAS-STING mechanism, synergizes with PD-1 antibodies, and consequently enhances T cell infiltration and cytokine secretion158. The inhibitory receptor mucin-containing molecule 3 (TIM-3), known for regulating extracellular DNA engulfment, influences chemokine expression, whereas depletion of cGAS and STING compromises this process, diminishing synergistic cytotoxicity with TIM-3 blockade159. Recent studies have highlighted the superiority of carbon ion irradiation (CIRT) to X-ray irradiation in inducing DNA damage and cGAS-STING activation. CIRT not only fosters immune cell infiltration but also amplifies immune checkpoint molecule expression, and slows melanoma growth when combined with PD-L1 inhibitors160. PRMT1 inhibitors activate cGAS/STING-dependent DNA sensing signals, in synergy with anti-PD-1 antibodies120. Zebularine, a demethylating agent, sensitizes the cGAS-STING pathway to DNA stimulation, in synergy with cGAMP and immune checkpoint blockade therapy, thus promoting immune cell infiltration161. Combined treatment with cGAMP and anti-CD47 monoclonal antibodies induces a systemic anti-tumor immune response via STING-IFN signaling and effectively inhibits tumor growth162. In a phase Ib multicenter study, the combination of ADU-S100 and the anti-PD-1 antibody spartalizumab has shown good safety profiles and high tolerance, with an overall efficacy rate greater than that of ADU-S100 monotherapy; this treatment therefore has promise for patients with late-stage solid tumors and lymphoma163.
Limitations of treatment strategies based on activating the cGAS-STING signaling pathway
The cGAS-STING signaling pathway is a crucial component of the immune system, and its activation induces robust anti-pathogen and anti-tumor immune responses. However, recent research has revealed a series of challenges and limitations in clinical trials, despite the promising potential of CDNs in preclinical studies, thus significantly affecting their widespread application and effective treatment of tumors.
First, whereas STING agonists have shown encouraging anti-tumor immune potential in preclinical studies, emerging evidence suggests that STING activation may have opposing effects in regulating anti-tumor immunity. The dual role of the cGAS-STING pathway is context dependent and intricately associated with the evolving TME. Its effects vary depending on factors such as hypoxia, nutrient availability, and the presence of immune and stromal cells. In some cases, sustained STING activation leads to an immunosuppressive TME, including the recruitment of MDSCs and Tregs78,164, impaired T cell proliferation, and diminished memory cell numbers165. The STING-IL-35 axis in B cells weakens NK cell proliferation and diminishes NK-driven anti-tumor responses72. Additionally, the release of cytosolic oxidized mtDNA in oral cancer induces IFN signaling via the cGAS-STING-TBK1 pathway, thus upregulating the expression of PD-L1 and indoleamine 2,3-dioxygenase 1, and inhibiting T cell activation166. In TNBC cells, STING-mediated NF-κB induces IL-6 expression and activates pSTAT3, thereby enhancing cell survival rates and PD-L1 expression167. Therefore, the bidirectional effects of the cGAS-STING pathway in cancer emphasize the need for a nuanced understanding of its regulation and function in different tumor contexts. Further research on the specific molecular mechanisms underlying this duality will be essential for unlocking the full therapeutic potential of cGAS-STING modulation in cancer treatment.
The use of STING agonists is also hampered by the non-specificity of drugs. Currently, almost all available STING agonists lack tumor specificity and may potentially activate STING non-specifically throughout the body, thus leading to “on-target off-tumor” toxicity; moreover, intravenous or intraperitoneal administration of STING agonists may trigger cytokine storms168. Therefore, identifying STING agonists specific to tumor tissues and optimizing dosages and timing to avoid excessive STING pathway activation will be essential to avoid worsening clinical outcomes. For example, positron emission tomography imaging of 18F-labeled STING agonists based on non-nucleotide MSA-2 has indicated improved affinity and specificity169. STING agonists conjugated with antibodies can be specifically delivered to sites expressing tumor-specific antigens170. Furthermore, Ding et al.171 have designed and synthesized a photo-caged STING agonist 2 with a tumor cell-targeting carbonic anhydrase inhibitor warhead, which is readily uncaged by blue light and releases the active STING agonist, thereby significantly activating the STING signal. These efforts have provided valuable insights for developing more tumor-specific drugs.
The adverse effects of STING agonists also limit their application. DNA-damaging agents such as radiation and anthracycline chemotherapy induce delayed cardiac inflammation, owing to the activation of the cGAS-STING-IFN-α/β signaling pathway after treatment172. In platinum-induced cardiac toxicity research, the STING-TNF-α-AP-1 axis triggers cardiomyocyte apoptosis173. In mice, genetic disruption of the cGAS-STING pathway inhibits DNA damage-induced cardiac inflammation, rescues late-stage cardiac function decline, and prevents cardiac events leading to death172. The STING pathway activated by mitochondrial stress and DNA leakage may lead to downstream neuroinflammation (such as neurodegenerative brain diseases)174. To alleviate the adverse effects of STING agonists, researchers have explored various administration routes and methods. For instance, Chen et al.175 have designed a small batch of esterase-activatable prodrugs based on the non-nucleotide STING agonist MSA-2, which stably binds liposome vesicles for intravenous administration. SAProsome liposomes improve delivery to the desired tumor and lymphatic compartments without causing significant systemic toxicity.
A recent study has highlighted the limitations of STING agonist treatment strategies. Li et al.176, using a novel single-cell RNA sequencing data analysis technique called ContactTracing, have confirmed that chronic activation of the STING pathway induced by chromosomal instability promotes downstream changes in cell signaling, which suppress effective anti-tumor immunity and facilitate cancer metastasis. This finding may explain why STING agonists have shown limited efficacy in clinical trials in patients with late-stage cancer. Additionally, compared with STING agonist development, the development of STING inhibitors remains in its infancy, and nodidate drugs are just entering clinical studies. Whether targeting STING inhibitors alone elicits sufficient therapeutic effects on tumors remains unclear, and the toxicity of these inhibitors is unknown; therefore, additional research is urgently needed177.
In summary, despite the potential of the cGAS-STING signaling pathway in anti-tumor immunity, CDNs face several challenges and limitations in clinical applications. In-depth research on the mechanisms of the cGAS-STING signaling in different types and stages of tumors, the development of tumor-specific STING agonists, and the establishment of effective biomarkers for patient selection is needed to overcome immunoresistance and activate immunogenic cell death pathways in cancer cells, thereby enabling the development of more effective treatment strategies.
Conflict of interest statement
No potential conflicts of interest are disclosed.
Author contributions
Conceived and designed the review: Ping Wang, Lan Fang.
Collected the data: Jiawen Zhang, Sihui Yu, Lan Fang, Qiao Peng.
Figure preparation: Jiawen Zhang, Sihui Yu.
Wrote the paper: Jiawen Zhang, Sihui Yu.
Acknowledgments
We appreciate Dr. Hongqi Teng for revising and improving the English language of this manuscript.
Footnotes
↵*These authors contributed equally to this work.
- Received November 13, 2023.
- Accepted December 14, 2023.
- Copyright: © 2024, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.
- 21.↵
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.↵
- 174.↵
- 175.↵
- 176.↵
- 177.↵
In this issue






{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- cGAS-STING signaling pathway
- The cGAS-STING pathway in cancer biology
- Strategies for harnessing the cGAS-STING pathway in cancer therapy
- Limitations of treatment strategies based on activating the cGAS-STING signaling pathway
- Conflict of interest statement
- Author contributions
- Acknowledgments
- Footnotes
- References
- Figures & Data
- References
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.