

Abstract
In oncolytic virus (OV) therapy, a critical component of tumor immunotherapy, viruses selectively infect, replicate within, and eventually destroy tumor cells. Simultaneously, this therapy activates immune responses and mobilizes immune cells, thereby eliminating residual or distant cancer cells. However, because of OVs’ high immunogenicity and immune clearance during circulation, their clinical applications are currently limited to intratumoral injections, and their use is severely restricted. In recent years, numerous studies have used nanomaterials to modify OVs to decrease virulence and increase safety for intravenous injection. The most commonly used nanomaterials for modifying OVs are liposomes, polymers, and albumin, because of their biosafety, practicability, and effectiveness. The aim of this review is to summarize progress in the use of these nanomaterials in preclinical experiments to modify OVs and to discuss the challenges encountered from basic research to clinical application.
keywords
Introduction
In the past decade, tumor immunotherapy has made substantial progress in restarting and maintaining the tumor immune cycle, and activating the body’s anti-tumor response1. In oncolytic virus (OV) therapy, an essential branch of tumor immunotherapy, viruses replicate in and lyse tumor cells through various regulatory mechanisms, without affecting the growth of normal cells2–4. Currently, dozens of OV drugs have been developed for tumor treatment, including adenovirus (Ad), vaccinia virus, measles virus, poliovirus, herpes virus, retrovirus, reovirus, parvovirus H1, Newcastle disease virus, and coxsackievirus5–8. The administration methods have included primarily intratumoral injection, intravenous injection, intraperitoneal delivery, and hepatic artery infusion2,6.
Four OVs have been approved for clinical tumor treatment: Rigvir, T-vec, H101, and G47Δ9–11. Although the currently marketed products are administered locally, the OV administration route with the highest efficiency and safety in clinical trials is intratumoral injection, a route used primarily for surface tumors or localized tumors6,12. Clinically, OVs are used primarily to treat malignant tumors after failure of second- and third-line therapy; these tumors often exhibit metastases13. Although OVs injected into tumors are not transported to metastatic tumors, they can lead to the control of metastatic tumors (“distal effect”) by activating host antitumor immunity14,15. However, because therapeutic efficacy is limited, metastatic tumors remain difficult to cure. In addition, intratumoral injection has several insurmountable drawbacks: 1) the tumor tissue has highly elevated density and pressure, both of which limit successful entry of OVs into tumors4,16; 2) when a tumor grows deep in the human body, intratumoral injection is impossible17; 3) because of the abundant blood vessels in tumors, intratumoral injection may cause tumor rupture and enormous hemorrhage, and risks of tumor tissue shedding and metastasis may be present18; and 4) compliance is poor among patients who require industrial drug administration, particularly long-term medication3.
OV-associated clinical trials are increasingly testing various modes of administration, particularly intravenous injection19. However, the existing intravenous drugs still have many problems, such as poor drug stability, low targeting efficiency, low solubility, and adverse reactions20–22. Recent developments have led to increased use of nanotechnology in medicine, pharmacy, and biology. Nanoparticle delivery systems significantly increase the water solubility of drugs; prolong the half-life; and improve the loading of preparations, targeting, and biological safety23. In solid tumors, certain macromolecular substances are likely to penetrate into the tumor tissue and remain there for longer periods than in normal tissue, because of tumors’ abundant blood vessels, large vessel wall space, and lack of lymphatic circulation. This phenomenon has been termed the enhanced permeability and retention (EPR) effect.
In recent years, numerous preclinical studies have used various nanomaterials for drug delivery and have yielded promising results24,25. However, only several nanostructures have been approved for the clinical market to date26. On the one hand, this lack of approvals is because mice are the most commonly used animal models, and mouse solid tumors somewhat differ from those in humans. On the other hand, clinical translation inevitably requires large-scale production of anticancer drugs, and the price, safety, stability, and reproducibility of drug preparations of nanomaterials are critical27,28. To date, many nanoparticle-based medicines, primarily lipid, polymers, and albumin nanostructures, have been successfully applied in the clinical treatment of tumors29 (Table 1).
Clinically approved liposomes, polymers, and albumin drugs
In this review, we describe the advantages and disadvantages of lipids, polymers, and albumins as delivery vehicles (Table 2). We then describe related studies on the delivery of OVs by using lipid, polymers, and albumin (Table 3), and finally discuss their respective application challenges in the clinical treatment of tumors.
Advantages and limitations of liposomes, polymers, and albumin in drug delivery
Drug delivery of OVs by liposomes, polymers, and albumin
Liposomes
In 1965, Zahednezhad et al.30 first proposed the idea of liposomes. In the 1970s, liposomes were widely recognized as a drug carriers31. In 1988, a liposome gel containing econazole was successfully marketed32. In 1990, injectable amphotericin B liposomes were listed in Europe33. Shortly thereafter, adriamycin liposomes were launched as the first cancer treatment drug liposome product in history. Currently, several liposome preparations have been marketed and widely used in clinical practice34, including daunorubicin liposomes, cytarabine liposomes, paclitaxel liposomes, and 5-fluorouracil heterophasic lipid plastids. Lipid materials have become the most widely used drug carriers, because of their advantages in encapsulating and transporting drugs35 (Figure 1). Liposomes are most commonly made of phospholipids and cholesterol, which are endogenous substances existing in organisms, with good histocompatibility and no immunogenicity36. The phospholipid molecules in the human body’s lipid bilayer each have a hydrophilic head made of phosphate and a hydrophobic tail made of 2 chain fatty acids, which confer both hydrophilicity and hydrophobicity37. Their internal hydrophilic environment makes liposomes suitable for encapsulating highly water-soluble drugs and gene fragments, whereas the middle of the bilayer, where the phospholipid tails are located, is a suitable location for storing and continuously releasing lipophilic drugs.

Schematic diagram of liposome encapsulated oncolytic virus.
Advantages and disadvantages of lipids as nanomaterials for drug delivery
The reasons for choosing liposomes as delivery carriers are as follows. 1) Dose control: Antitumor therapy can be administered at a dose that is more acceptable and with less systemic toxicity by using liposome-loaded medicines38. 2) Enhancing therapeutic effects: When liposomes are modified with hydrophilic polymers, such as polyethylene glycol (PEG), they are less likely to be removed by the liver’s and spleen’s mononuclear phagocyte system (MPS), thus effectively extending the liposomes’ in vivo circulation time and consequently increasing tumor enrichment. These aspects enable repeated dosing with liposome drug delivery systems39,40 (Figure 2). 3) Target delivery: After binding tumor cell target proteins, ligand modified liposomes enter cells through receptor mediated endocytosis41. The advent of liposomes responsive to stimuli including temperature, pH, and hypoxia has accelerated the application of nanomedicines for site-specific delivery42. 4) Arbitrary modification: Through internal or external physical, chemical, and biological stimuli, biological differences between tumor cells and normal tissues can be targeted. Modification of nanodrug carriers or molecular structures enables control of drug release timing and release rates, and improves drug utilization. 5) Additional advantages: Lipid-mediated drug delivery provides unrestricted packaging capacity, relative ease of mass production, and carrier delivery with high copy numbers43.

Modifying liposomes with PEG avoids clearance by the mononuclear phagocytic system (MPS) of the liver and spleen. Synthesis scheme for PSar (A) and schematic illustration of PEG- and PSar-liposomes (B). Ex vivo imaging of PEG-liposomes (C) and PSar-liposomes (D) in Wistar rats after the first and second injections. Images were obtained 4 h after injection. Ex vivo emission intensity of organs from Wistar rats with PEG-liposomes (E) and PSar-liposomes (F) 4 h after the first and second injections. Each bar represents the mean ± SD (n = 4–5). Copyright © 2020 Elsevier B.V. Sar-NCA, sarcosine-N-carboxyanhydride; HATU, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate; DIEA, N,N-diisopropylethylamine; PSar, polysarcosine; HSPC, hydrogenated soya phosphatidyl choline.
However, despite the clear advantages of drug delivery via liposomes, several problems exist with this strategy: 1) Excessive volume: Liposome-encapsulated viruses may become too large (greater than 100 nm) to exploit the EPR effect44. 2) Contagiousness of OVs: Because the infectivity of oncolytic adenoviruses (OAs) (including telomere scanning) largely depends on expression of the coxsackie and adenovirus receptor (CAR) in cells, and the attachment of the viral capsid, including the attachment of fibrin and viral receptors to the cell surface, the original telomerase-specific oncolytic adenovirus (TelomeScan) cannot produce cytotoxic effects in cancer cells with low CAR expression levels. TelomeScan (10 or 20 MOI) significantly inhibits the infectivity of HCT116 cells in culture medium supplemented with CAR antibodies. The presence of CAR antibodies had little effect on the ability of liposome-encapsulated plasmid DNA of telomerase-specific OAs (TelomeScan) to express GFP (Lipo-pTS). Because the CAR infectivity of Lipo-pTS is independent of, and different from, the original TelomeScan, our findings suggest that Lipo-pTS has the potential to infect and achieve cytotoxic activity, even in cancer cells with limited CAR expression on the cell surface45. 3) Single structural component: The reticuloendothelial system eliminates liposomes that enter the body, and the single structural component of liposomes is too simple compared to the biologically active cell structure46. 4) Instability: The instability of liposomes is also a difficult problem to solve, and is usually associated with the oxidation and hydrolysis of liposomes, drug leakage, and even liposome fusion47. 5) Difficulty in achieving sterile operations: The sterilization process also limits the application of liposomes; because of the tendency of liposomes to deteriorate or degrade after sterilization, suitable and effective sterilization techniques are necessary to ensure sterility48.
Advances in lipid-encapsulated OVs
Antiviral antibodies have been found to exist in most of the population, and over 80% of adults possessing anti-Ad5 antibodies49. The first problem encountered with intravenous OVs is attack by neutralizing antibodies. As early as the 1990s, liposomes were used as carriers of OVs to ward off attacks from neutralizing antibodies. To fight against Ad-specific neutralizing antibodies (AdNABs), Katsuyuki et al.45 have studied the potential of TelomeScan plasmid DNA encapsulated in liposomes capable of expressing GFP (Lipo-pTS) to function as an oncolytic adenoviral agent for systemic delivery. In HCT116 colon cancer cells, lipo-pTS with a diameter of 40–50 nm has been found to have anti-tumor efficacy, as well as tumor specificity unrelated to CAR. In addition, intravenous lipo-pTS decreases the generation of AdNABs and retains high cytotoxicity even when AdNABs are present45. Fu et al.50 have prepared 3 different forms of HSV vectors: HSV capsids, complete viral particles, and purified viral DNA. All 3 types of HSV can easily produce contagious viruses when transfected into cultured cells, because intravenous liposome delivery of HSV vector DNA successfully avoid host immune neutralizing antibodies. Moreover, liposome-cloaked oncolytic Ad (oAd) conjugated to tumor-homing Escherichia coli BL21 (designated E. coli-lipo-oAd) has been found to enhance cancer immunotherapy51 (Figure 3).

Schematic representation of self-propelled bacterium vessels carrying OAs that can home to tumor lesions and enhance antitumor responses through bacterial–viral-augmented immune activation. Copyright © 2022 American Chemical Society. DC, dentritic cell.
In recent years, several studies have shown that liposome-encapsulated OVs significantly increase the antitumor ability of OVs. Among them, cationic liposomes has become a major topic. Kwon et al.52 have complexed negatively charged oncolytic Ad genomic DNA with cationic liposomes (DOTAP/DOPE) to generate pmT-d19/stTR+DOTAP/DOPE lipoplexes, which have been found to achieve comparable cell death to that induced by oAd after treatment with liposomes. Therefore, Ad genomic DNA in liposomes can efficiently generate active oncolytic Ad progeny. In addition, systemic administration of pmT-d19/ stTR + DOTAP/DOPE liposomes have been found to elicit an extremely effective antitumor response in vivo, resulting in a 90.5% and 92.4% lower tumor volume than observed after bare-colytic Ad or bare-colytic Ad genomic plasmid therapy, respectively. The antitumor effect of oncolytic Ad DNA liposomes is better than that of naked oAd and naked oAd genome plasmids53. Sakurai et al.54 have complexed reovirus with cationic liposomes as a transfection agent. Reovirus cationic liposome complexes (reoplex) and reovirus alone both showed equivalent levels of tumor cell killing efficacy in reovirus-sensitive tumor cells, whereas the degree of tumor cell killing activity in resistant tumor cells was more than 30% higher than that of reovirus alone. Receptor-mediated tumor cell death was successfully induced in tumor cells that had previously received cathepsin inhibitor treatment. Interferon (IFN)- and apoptotic gene mRNA levels markedly increased during repeated compounding. According to these findings, cationic liposomes effectively encourage the transfer of reoviruses to the cytoplasmic matrix and subsequent induction of apoptosis.
However, after entering the body, cationic liposomes absorb negatively charged serum proteins and consequently cause liposome nanomaterials to aggregate, thus significantly decreasing their ability to penetrate tumors via the EPR effect44. In eukaryotic cell membranes, anionic liposomes have the benefits of being less immunogenic and hazardous than cationic liposomes. In addition, anionic liposomes can undergo a phase change when calcium ions are present, thereby aiding in the electrostatic adsorption process through which liposomes encapsulate OVs55. In addition, Wang et al.56 have used the membrane hydration method to encapsulate M1 into soybean lecithin liposomes; these liposomes effectively prevented M1 neutralizing antibody from binding M1 without affecting viral infectivity, thereby blocking the immunogenicity of the virus and enhancing its killing effect on colon cancer (LoVo) and human hepatoma (Hep 3B) cells. Zhong et al.55,57 have reported a calcium-induced phase transition approach to encapsulate Ad5 in anionic liposomes (Ad5-AL). Gene expression was 6-fold higher in adenocarcinoma cells infected with Ad5-AL than in cells infected with the bare virus. In addition, an antibody neutralization experiment revealed that Ad5-AL was more efficient than the complex of Ad5 and cationic liposomes (Ad5-CL) in shielding Ad5 from neutralization, because it inhibited both naked Ad5 and Ad5-CL at higher dilutions. This finding may have 2 explanations. First, the Ad5-AL group had higher gene expression; second, a Ca2+ chelator increased Ad5 uptake and gene expression. Thus, these findings demonstrate that anionic encapsulation in liposomes enhances the transfection efficiency of Ad5 and significantly increases gene expression in mouse airway tissues when delivered by intratracheal instillation in vivo58.
Application prospects
Broad applications and substantial development possibilities exist for OV encapsulation and delivery to tumor locations by using lipids. Modified lipid drug delivery systems enable a range of functions, such as boosting therapeutic efficacy, lowering systemic adverse responses, avoiding drug resistance, destroying tumor stem cells, and penetrating biological barriers. Therefore, these lipids are worthy of research and development. The theoretical study of lipids is becoming more complex, and research on laboratory lipid preparation techniques is maturing. Lipids can be used in a variety of methods and applications, whether systemic or topical, to enable targeted drug delivery that effectively deposits drugs at lesion sites, thus achieving better effects while minimizing unwanted responses. However, the industrialization of lipid nanoparticle drug delivery systems continues to pose barriers to their advancement, such as onerous quality control procedures and exorbitant pricing for membrane components including injection-grade phospholipids. Lipid drug delivery systems will have excellent development prospects as science advances, and pharmaceutical devices and technologies are regularly updated.
Polymers
A polymer is a substance with a high molecular weight that is composed of several identical and simple structural units linked repeatedly by covalent bonds. Because of their ease of synthesis, polymer nanostructures have a wide range of applications in multiple aspects of nanomedicine. First, polymer nanomaterials can be directly used as nanomedicines for the treatment of diseases; the most famous example is Copaxone®, a synthetic peptide compound. In 1996, Copaxone® was first approved for marketing in Israel and in the same year was approved by the US FDA for the treatment of multiple sclerosis59. However, polymer nanomaterials are also commonly used to deliver drugs. Polymers and drugs are linked by chemical bonds. After entering the body, chemical bonds are broken by endogenous or exogenous changes, and the drug is released at the target site (Figure 4). In the 1960s and 1970s, polymers were developed for biodegradable sutures. Since then, because of polymers’ excellent degradability and bioavailability, the field of drug delivery has flourished60. In recent years, polymer nanomaterials with targeted controllable release have been used to encapsulate drugs; in vitro laboratory research and increasing clinical trials have already led to the approval of several products on the market61. Polymers are applied in 2 main drug types. The first type is polymer-drug conjugates, which are used to increase the bioavailability and half-life of drugs. One representative nanomaterial is PEG, in the drug PLEGRIDY®, which was approved by the FDA in August 2014 for the treatment of adults with relapsing-remitting multiple sclerosis62. Another representative drug, adynovate, was approved in 2015 for the treatment of pediatric hemophilia A in patients under 12 years of age63. The second type of polymeric drug uses the degradability of polymers to achieve controlled drug release. A representative nanoparticle is poly-(lactic-co-glycolic acid) (PLGA), and the representative drug Eligard® is commonly used to treat advanced prostate cancer64.
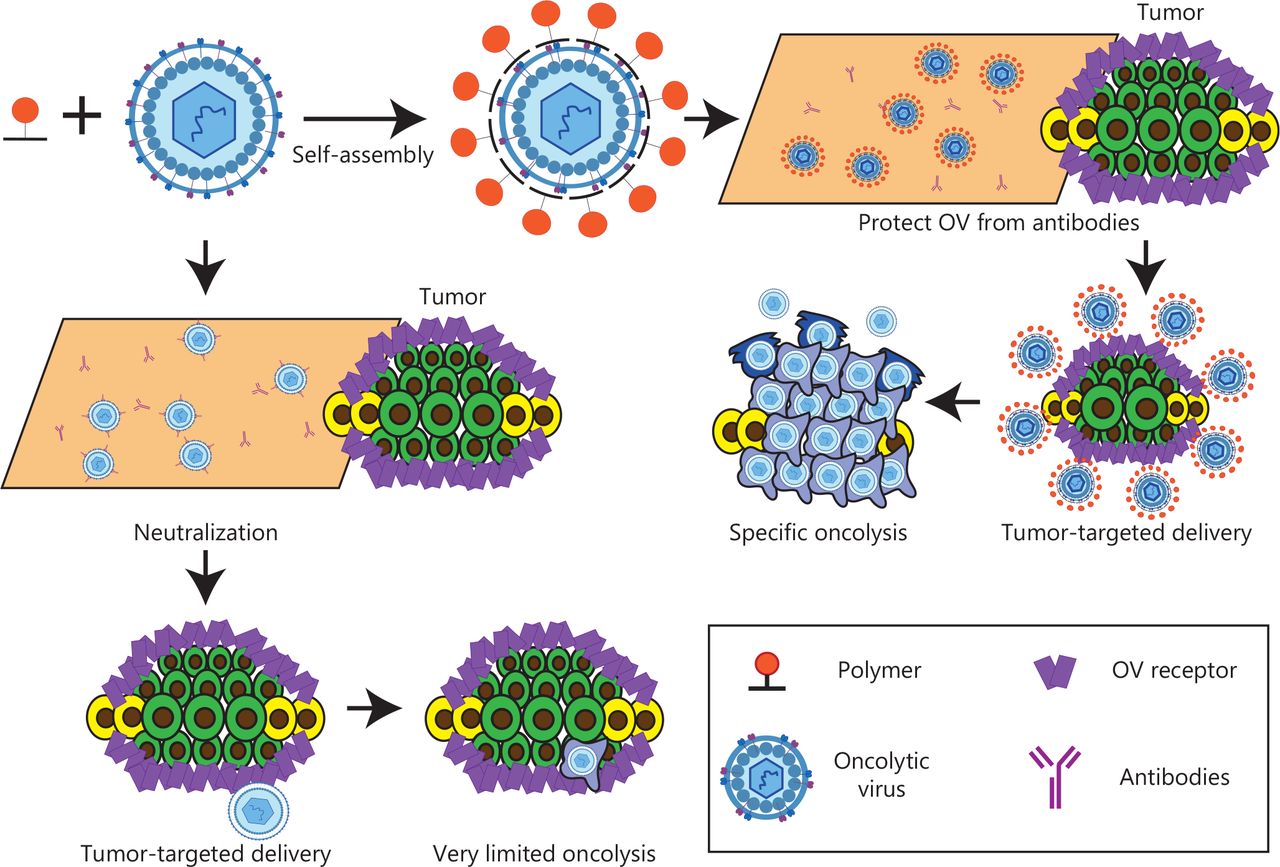
Schematic diagram of polymer-encapsulated oncolytic virus.
Advantages and disadvantages of polymers as nanomaterials for drug delivery
Overall, the main advantage of using polymers as drug carriers is their biodegradability. Biodegradable polymer drug carriers can be broken down in the body to produce nontoxic natural byproducts, such as carbon dioxide and water. Biodegradable polymers can divided into 2 categories, synthetic and natural, each with its own advantages and disadvantages65. Synthetic polymers, for example, are characterized by their precisely controlled chemical structures and biological inertness. Therefore, they exhibit excellent chemical versatility and batch-to-batch uniformity66. However, the development of synthetic polymers is also limited by their biological inertness67. In addition, an essential topic in the field of polymer science is enhancing the interaction between polymers and target cells, mainly by incorporating functional materials into the structures of synthetic polymers68. In contrast, unlike natural polymers, synthetic polymers can be hydrolyzed without a need for enzymes69. Natural polymers have 3 major characteristics: strong biological activity, excellent biocompatibility, and degradation by proteolysis through cell activation70. However, higher biological activity leads to a stronger immunogenic response. Furthermore, natural polymers are obtained from natural resources, and their chemical structures are difficult to purify, characterize and identify accurately, thus leading to batch-to-batch variability. These 2 major issues have hindered further development71.
Another substantial advantage of polymer drug carriers is the controlled release of drugs72. The endogenous controlled release mechanism of polymers can be summarized as follows: 1) drug diffusion through water-filled pores; 2) osmotic pumps; 3) drug diffusion through the polymer matrix; and 4) substantial erosion. The exogenous controlled release mechanism triggers release through structural changes caused by endogenous and exogenous stimuli (for example, polymer surface layer shedding, charge conversion, and degradation). Stimuli include temperature, light, specific biomarkers (such as urea and glucose), redox potential, and any combination thereof (such as pH/temperature, pH/magnetic field, and so on)73. Polymer gels and polymer micelles are common vectors for achieving exogenous controlled release responses with drug delivery systems.
Polymer-based drug carriers also have advantages, such as improved stability, prolonged circulation time, diminished adverse effects, adjusted physical and chemical properties, and versatility (with payloads as diverse as anticancer drugs and gene therapy reagents)74. Although polymers have numerous advantages as drug carriers, they still face at least 6 major obstacles that must be overcome to enable extensive clinical application: 1) drug loading is limited in comparison to that of more common drug carriers, such as liposomes; 2) the drug’s tumor accumulation rate is most likely to be modest, because of subpar vascular extravasation and other factors; 3) tumor penetration is weak (the tumor has dense extracellular matrix and elevated interstitial fluid pressure); 4) partial polymer materials decrease the transfection rate of drugs, such as PEG; 5) the cytoplasmic drug release efficiency is low, because of encapsulation in the acidic endosome/lysosome compartment; and 6) for some natural polymers, determining the material’s composition and assessing its biological effects are difficult, thus hindering development of drug delivery composite nanostructures75–77.
Advances in polymer-encapsulated OVs
PEG
PEG, a hydrophilic linear polymer, has biocompatible and nonimmunogenic characteristics78. These properties make PEG the polymer of choice for the delivery of OVs79 (Figure 5). The amount of surface modification and the molecular weight of PEG significantly affect the amount of PEGylation that can be achieved for OVs80,81. Ad modification with low molecular weight PEG (2–5 kDa), for instance, does not decrease viral accumulation in the liver80. In contrast, liver accumulation decreases when Ad is modified with slightly higher molecular weight PEG (20–35 kDa)82. Accelerated blood clearance (sometimes referred to as the “ABC phenomenon”) is a unexpected immunogenic reaction seen in PEG conjugates that causes rapid clearance of PEG-based nanocarriers. After repeated administration, the widely documented ABC phenomenon decreases the potency of PEG conjugates and nanocarriers. C activation-associated pseudoallergy, another unanticipated immune response, significantly decreases the safety of PEG-based nanocarriers and has been associated with a decline in the efficacy of PEG-based therapy in clinical trials. One important structural component affecting immunological safety is PEG length. Long and short chain PEG conjugates are relatively more likely to cause the ABC phenomenon, because this effect is biphasic. PEG density exhibits a biphasic effect similar to that of PEG length. However, PEGs with diminished ABC phenomenon have been identified among lower and higher density PEGs. Yao et al.83 have reported that PEGylation with 45% coverage using 20 kDa PEG, targeting reactive amines, yields the optimal PEGylation outcome for Ad.
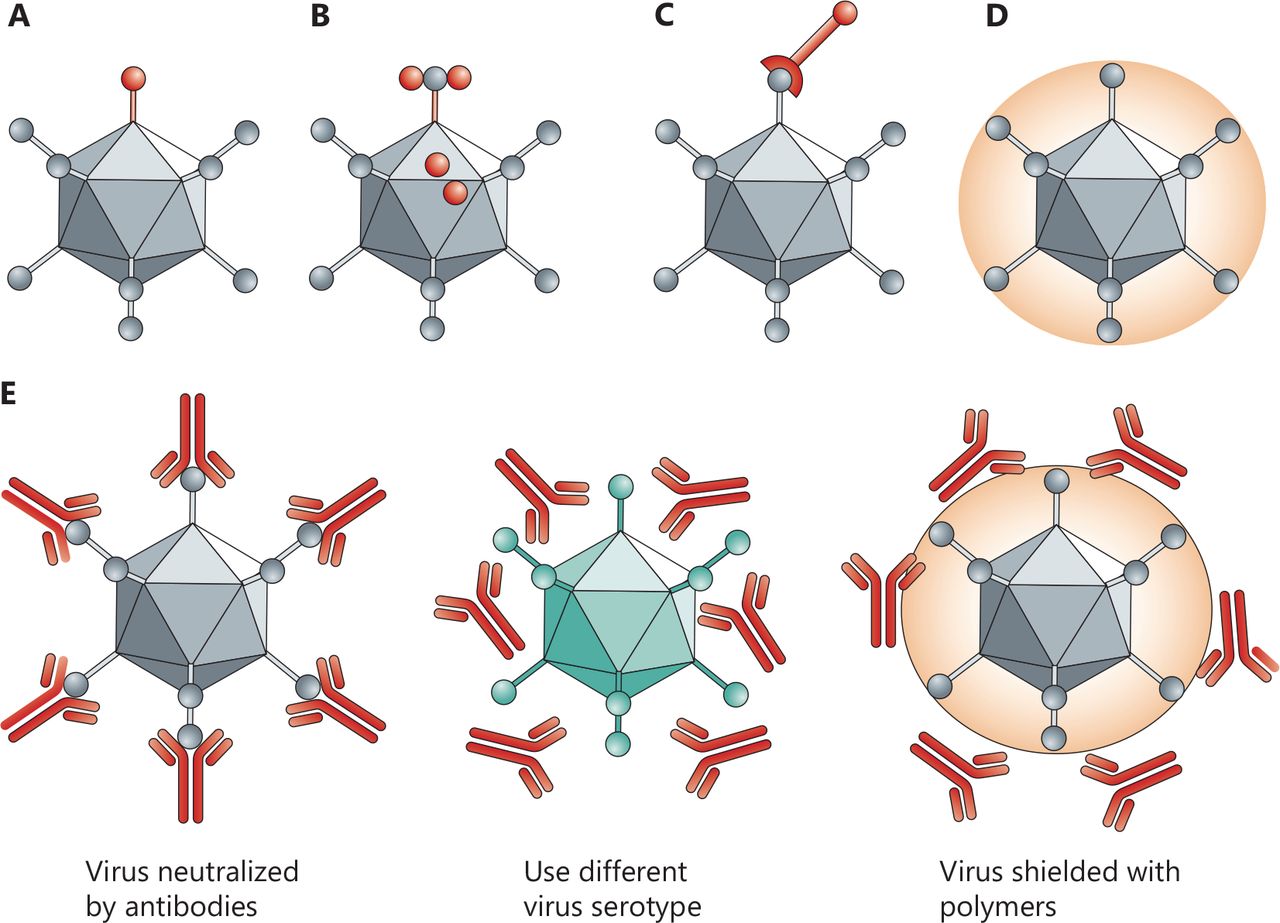
Biochemical vs. genetic modification of viruses. In genetic retargeting, an exogenous ligand is fused to, or replaces, the normal receptor-binding protein of the virus. (A, B) Viral particles can be modified chemically. (C–E) Shielding the virus against antibodies. Pre-existing neutralizing antibodies in humans can interfere with efficacy. Changing virus serotypes and coating particles with shielding polymers can address the neutralizing-antibody problem. Copyright © 1969, Springer Nature.
O’Riordan et al.84 have demonstrated that PEGylated Ad is more resistant to inactivation by AdNABs than bare Ad. The covalent bond between PEG and the adenovirus surface is achieved primarily through the use of activated PEG trimethyl monomethoxy PEG, which preferentially binds lysine residues in an ε amino terminal reaction. The components in the capsid that trigger neutralizing immune responses—i.e., hexanone, fiber, and pentosan bases—are also the main targets of PEG formation, thus enabling PEG to effectively protect capsid proteins. Furthermore, Croyle et al.85 have shown that PEGylation of Ad successfully avoids NABs. However, because of the tendency of PEGylated Ad to accumulate and precipitate, these studies are currently applicable only to intratracheal injection. Wilson et al.86 have demonstrated that systemically administered naked Ad results in greater adaptive immune responses than PEGylated Ad. Specifically, bare Ad induces greater Ad-specific NAB production than PEGylated Ad. PEGylation of Ad decreases interleukin (IL)-6 levels in the serum after systemic delivery, thus blunting the development of innate antiviral immune responses87–89. PEGylated Ad markedly decreases hepatotoxicity and innate antiviral immune responses, but this shielding may diminish oncolytic efficacy after intravenous injection therapy90.
Despite the substantial benefits of PEGylation in improving systemic delivery of Ad, removing Ad’s ability to attach to CARs has been shown to weaken several essential components that support its higher transduction efficiency91. Ad undergoes a two-step process for cellular entry. Initially, Ad fibers bind CAR, and this is followed by engagement of cellular integrins (αvβ3 and αvβ5) and arginine glycine aspartate motifs (RGD) on the penton base of Ad. Additionally, the interaction of Ad5 vector particles with plasma proteins, such as coagulation factor X (FX), which binds the main Ad5 capsid protein hexon, significantly affects the distribution of Ad5 vector particles in living organisms92. Interestingly, PEGylated Ad decreases cellular internalization and hampers virion trafficking to the nucleus, with assistance from microtubules93. Therefore, additional investigations are required to address the restrictions of PEGylated Ad. Choi et al.94 have used PEG-poly(N-[N-(2-aminoethyl)-2-aminoethyl-2-aminoethyl]-L-glutamic acid copolymers (PEG-PLNG) to create a sequence of 6 biocompatible polymers, each with a distinct PEG molecular weight and amine group count (2 or 5). In general, the relationship between the polymers’ surface charge and size and amine content was inverse. The need for more amine groups within the polymer was demonstrated by the higher transduction efficiencies observed for Ad nanocomplexes made of PEG-PNLG variants with 5 amine groups in both CAR-positive and CAR-negative cells than Ad complexes made of PEG-PLNG with 2 amine groups94.
PLGA
PLGA, a copolymer comprising lactic acid and glycolic acid, is recognized as a biocompatible and biodegradable material95. Commonly referred to as a “smart polymer,” PLGA first found application in the development of absorbable surgical filaments during the early 1970s96. Over the past few decades, its utility has expanded, and it is currently among the most successful drug delivery systems, because of its remarkable properties. Its advantages include biocompatibility, sustained release capability, non-toxicity, non-immunogenicity, and an ability to accommodate a wide range of drugs with polymer-friendly adaptability97–99. Notably, the hydrolysis of PLGA yields endogenous compounds that are easily metabolized. Furthermore, PLGAs can be processed into amorphous or crystalline forms that vary in shape and size, thereby facilitating the encapsulation of diverse molecules, both hydrophobic and hydrophilic100,101. Badrinath et al.102 have embedded a cancer-favoring oncolytic vaccinia virus (CVV) on PLGA nanofibrous membranes and compared the therapeutic effects of empty PLGA nanofibrous membranes and CVV-PLGA nanofibrous membranes. Within 48 hours, anticancer activity of CVV was observed, on the basis of continuous CVV release from the PLGA nanofiber membrane, while the anticancer activity of CVV is correctly preserved during the embedding process. Studies using tumor xenografts in living organisms demonstrated that the CVV liberated from the PLGA nanofiber membrane was effectively transported into tumor tissue. Therefore, these findings indicate that although the membrane itself does not effectively restrict the growth of tumor volume, the PLGA nanofiber membrane implanted with CVV does (Figure 6).

CVV-PLGA nanofiber. (A) Diagrammatic sketch of the use of CVV-PLGA in tumor therapy. (B–E) Effects of treatment with CVV-PLGA on CT26 tumors in BALB/c nude mice. Copyright © 2018 Elsevier B.V. H&E, hematoxylin and eosin; GFP, green fluorescent protein.
Cationic polymers
Cationic polymers are promising carriers for viral gene delivery. Through ionic complexation, these polymers form nanoscale assemblies with genetic materials103–105. Integration of cationic polymers with viral vectors has been used to bolster their gene transfer efficiency. After complex formation, the surface charge of the therapeutic agent shifts from negative to positive, thus facilitating heightened interactions with anionic cell membranes and promoting cellular uptake of the therapeutic drug106.
Arginine
Recently, gene delivery techniques using arginine-grafted biorepeatable polymers (ABPs) have been created107. Because arginine is typically present in protein transduction domains and cell-penetrating peptides, adding it to Ads can markedly increase their transduction efficacy108. ABP also has reducible disulfide bonds in its backbone, which enable it to biodegrade by shrinking the cytoplasm, thus effectively releasing Ads from the multiplex and decreasing cytotoxicity. ABP-coated Ad complexes have been shown to have diminished toxicity and improved transduction efficiency in both CAR-positive and CAR-negative cells. Additionally, unlike bare Ad, the ABP-coated Ad complex’s activity is not considerably decreased by serum, and innate immunological reactions and hepatotoxicity are both greatly mitigated. Decreased liver sequestration and increased anticancer effectiveness have been observed when cationic polymers or other hydrophilic polymers (such as PEG) are chemically coupled109.
pHPMA
pHPMA(poly-[N-(2-hydroxypropyl) methacrylamide]), another cationic polymer, has been extensively used for conjugation with OVs. Wang et al.110 have synthesized a series of pendant cationic oligolysine with varying lengths (K5, K10, and K15) through charge interactions for coating Ad5. Among these formulations, pHK10 exhibited superior transduction efficiency to the other 2 variants. Transduction tests conducted in CAR deficient Chinese hamster ovary cells indicated that pHK10 Ad5 virus, compared with Ad5, increased the transduction efficiency of cell entry. The cationic pHK10 formulation defeated Ad dependent CAR mediated cell entry, a finding attributed to the oligosaccharides found in HPMA. The main internalization pathway of polymer coated Ad5 was demonstrated to redirect from CAR to sulfated proteoglycans. Therefore, this straightforward viral modification technique may potentially provide a remedy for enhancing Ad5 conversion to a variety of CAR negative cell types.
To promote effective liver detoxification and decrease Ad neutralization by NABs, Fisher et al.111 have attached pHPMA to the Ad surface. Approximately 74% of the amine groups on Ad were altered by pHPMA, according to in vitro research. The Ad interaction with NABs decreased by almost 80% as a result of these changes. The ability of pHPMA to stop hepatic sequestration after intravenous delivery of Ads was highlighted by the observation that the liver transgenic expression levels after intravenous administration of pHPMA-modified Ads were 10,000 times lower than those of naked virions112–115.
Prill et al.116 have used pHPMA polymers activated with maleimide groups or pyridyldithio groups to examine the effects of coating Ad with a “bioresponsive” shield. According to their research, the addition of an irreversible coating affected how the virus was transported inside the cell to the nucleus. The bioactive covering did, however, eventually permit particle movement. The effects of the charge of Ad’s reactive polymer coating were then examined in vivo, and a positively charged reactive polymer coating was found to increase the likelihood of liver infection.
Carlisle et al.117 have applied a coating to Ads by using pHMPA-based polymers with bioreactive side chains containing amine-reactive TT groups and acid-labile hydrazone. In vivo tests showed a 50-fold greater circulation half-life and 8,000-fold lower hepatic accumulation than observed with naked Ad. Through ultrasound-assisted delivery, the tumor accumulation of the coated virus was improved, owing to its favorable circulatory profile. Combination of polymer-coated Ad, gas bubbles, and focused ultrasound enhances tumor infection by at least 30 times. In addition, the infectivity was 40-fold greater than that observed in controls without ultrasound, at distances more than 100 mm from the vasculature.
Furthermore, Carlisle et al.118 have examined how pHPMA coating affects other adeno-associated viruses (AAV) beyond Ad5. They discovered that using an established, effective adenovirus encapsulation method to combine reactive HPMA copolymers with AAV5 did not decrease AAV5 infection, because the presence of free polymers in fact promoted viral infection. An additional study has revealed that the increased activity induced by free polymers is mediated through typical infection mechanisms and has some resistance to neutralizing antibodies. While this observation was essentially an experimental artefact, the phenomenon might be applied to stop viruses from spreading locally in the environment.
PEI
Polyetherimide (PEI), which has remarkable cellular uptake and endosomal escape properties, has long been regarded as the gold standard for gene delivery119. However, the cytotoxicity of 25 kDa PEI substantially restricts its therapeutic use120–122. A polydegradable, bioreductible, core-crosslinked PEI (rPEI) copolymer with different molecular weights (16 and 32 kDa) has been created by Choi et al. and coated on the surfaces of OAs. Ad complexed with 16 kDa rPEI has shown greater transduction efficiency and cancer cell killing efficiency than naked Ad in both CAR-positive and CAR-negative cells123. A variety of cancer cells can be treated, because this increased anticancer cytotoxicity is more readily apparent in CAR-negative MCF7 cells. A549 and HT1080 cancer cells treated with Ad/16 kDa rPEI also display considerably lower Met and vascular endothelial growth factor (VEGF) expression than naked Ad or Ad/25 kDa PEI. These results indicate that the combination of shMet expressing oncolytic Ad and biodegradable co-crosslinked PEI can serve as an effective and safe cancer gene therapy. A polydegradable cationic carrier based on the mPEG-PEI-g-Arg-S-Arg-S-Arg-g-PEI-mPEG (Ad/PPSA) copolymer was developed by Jung et al.124 for coating Ad. The measurement of Ad/PPSA particle size demonstrated that the overall size and cationic charge increased with increasing polymer concentration. Complexing Ad with PPSA resulted in a 2.24-fold increase in anticancer activity in MCF7 tumor xenografts. Intravenous injection of DWP418/PPSA led to lower innate immune responses than naked Ad, because it released interleukin-6 cytokines into the serum. DWP418/PPSA’s vector targeting to CAR negative and positive cells, and its improved anti-tumor effects, indicated the promise of this tool in cancer gene therapy. Nosaki et al.125 have gradually added cationic PEI and anionic chondroitin sulfate to form an oncolytic measles virus (MV) multimolecule. The polymer-coated virus displayed greater oncolytic activity in vitro than the bare the genetically engineered measles virus (MV-NPL) in the presence of anti-MV neutralizing antibodies. Additionally, animals treated with the polymer-coated MV-NPL showed greater complement-dependent cytotoxicity and antitumor activity than mice treated with the bare virus. This new polymer-coated MV-NPL is anticipated to contribute to clinical cancer treatment in the future126.
Passive-targeted polymer
Complexation with polymers containing targeting moieties can be used to improve preferential tumor accumulation of systemically delivered OVs. Relaxin (RLX)-expressing oncolytic Ads (oAd/RLX), which break down the dense tumor extracellular matrix in highly desmoplastic pancreatic cancer, have been complexed with biodegradable polymer [poly(ethyleneimine)-conjugated poly(CBA-DAH); PCDP]. This combination allows human bone marrow-derived mesenchymal stromal cells to integrate more effectively than weakly loaded naked Ads127. Ad/RLX-PCDP-loaded human bone marrow-derived mesenchymal stromal cells have shown effective Ad uptake and proliferation after systemic treatment. In an orthotopic pancreatic tumor model, our method achieved stronger anticancer effects than observed with bare Ad by increasing tumor cell death due to apoptosis, as well as decreasing the tumor extracellular matrix.
Using a U87 tumor xenograft mouse model, Choi et al.128 have created the pH-sensitive block copolymer, methoxy poly(ethylene glycol)-b-poly(l-histidine-co-l-phenylalanine) (PEG-b-PHF) to target the acidic tumor microenvironment and limit tumor growth. They have investigated tumor-targeting capabilities by complexing pH-sensitive polymers encoding an oAd transcriptional inhibitor of the VEGF promoter (KOX). The cancer cell killing effects of KOX/PEG-b-PHF (pH 6.4) were greater than those of naked KOX and KOX/PEG-b-PHF (pH 7.4), thus suggesting more effective antitumor activity. Inspired by this method, the authors produce a PEG-b-pHis/Ad complex by physically complexed Ad with a pH-sensitive block copolymer, methoxy poly(ethylene glycol)-b-poly(l-histidine) (mPEG-b-pHis) under acidic pH conditions in tumor tissue129. The PEG-b-pHis/Ad complex considerably outperformed naked Ad in terms of conduction efficiency at pH 6.4, and showed significantly greater therapeutic efficacy in both CAR-positive and CAR-negative tumor types.
With the goal of covering the Ad surface, Moon et al.130 have created a pH-sensitive and bio-reducible polymer (PPCBA). The Ad-PPCBA complex responded to pH, displaying greater cellular absorption at pH 6.0 and improved cellular uptake in both CAR-positive and CAR-negative cancer cells at pH 7.4. Importantly, in a human xenograft tumor model, intratumoral and intravenous treatment with Ad-PPCBA nanocomplex expressing VEGF-specific shRNA resulted in a 3-fold greater anticancer effects than naked Ad, with lower levels of systemic toxicity. According to these results, adding bioreducible linkers improves pH sensitivity and increases envelope OV release within the intracellular space of cancer cells.
Tumor-targeted polymers
In addition to passive targeting, the incorporation of tumor-targeting motifs actively targets several receptors that are overexpressed in tumor tissue. The first research demonstrating successful tumor-targeting OVs used a folate (FA)-anchored Ad/chitosan-PEG-FA nanocomplex created by Park et al.131. To prevent impairment of Ad function, the ionically cross-linked chitosan layer on the Ad surface offers a site for chemical coupling with PEG and subsequent binding to FA (targeting moiety at the end of heterofunctional PEG). In comparison to bare Ad, PEGylation via the chitosan amine group caused a lower immunological response and an extended plasma circulation half-life. Ad/chitosan-PEG-FA nanocomplexes, compared with bare Ad, resulted in 75.3% lower Ad-specific NAB synthesis in mice; moreover, the hepatic accumulation was 378-fold lower132. Additionally, after subcutaneous administration of a systemic dose of Ad/chitosan-PEG-FA nanocomplex, mice with KB tumors expressing the folate receptor (FR) showed a 285-fold greater intratumoral viral load than observed with bare Ad. These results suggest that, by attaching polymers with appropriately localized moieties to the surface of OVs, tailored distribution of OVs can be accomplished without compromising viral infectivity.
Dendrimers made of polyamidoamine (PAMAM) have good water solubility, are not immunogenic, and have surface functional groups that are easily modified with drugs and targeted ligands. PEGylated PAMAM dendrimers (PPE) and EGFR-specific Erbitux antibodies have been used by Yoon et al.133 to create complexes with Ad. Because of steric hindrance, complexation with PEG based PAMAM (PP) dendrimers inhibits Ad transduction. In an EGFR-positive orthotopic lung tumor model, systemic injection of OA/DCN-shMet/PPE has been found to result in a remarkable 14.9-fold suppression of tumor growth with respect to naked Ad, thus causing total tumor regression in 66% of treated animals. A 290-fold increase in viral genomes found in the blood after 24 hours by pharmacokinetic profiling suggests that ErbB coupled and PEG functionalized PAMAM dendrimers can effectively mask the capsid of Ad and delay the effective internalization of oncolytic Ad into EGFR positive tumors, while decreasing the toxicity of systemic administration of nude oncolytic Ad134.
Different polymers
Biodegradable poly (cystaminoacrylamide-diaminohexane) [poly (CBA-DAH)] (CD) has been studied as a polymeric vehicle for oncolytic Ad dispersion. When Ad/CD-PEG-RGD is physically compounded with RGD peptide-coupled CD polymer (CD-PEG-RGD), the pathogenic effect of oncolytic Ad releasing shRNA against IL-8 has been found to be dramatically and dose-dependently amplified, as compared with the cell specificity of naked Ad in cancer cells. RGD-conjugated polymers also send Ads to cells that express certain integrins, regardless of CAR status135. Ad/CD-PEG-RGD treatment decreases the expression of IL-8 and VEGF while increasing apoptosis in the cells.
Garofalo et al.136 have used galactosylated polymers as carriers for systemic delivery of OAs in hepatocellular carcinoma cell lines. The results have demonstrated that the use of computational polymers for coating and targeting is a safe and effective therapeutic strategy with minimal adverse effects. These findings provide a basis for future studies using OV complexed with polymers for specific and more efficient targeting of hepatocellular carcinoma.
Hill et al.137 have coated vaccinia virus (VV) with an amphiphilic polymer, and attached an antibody-targeting anti-mucin-1 (aMUC1), thus yielding aMUC1-PCVV (polymer-coated VV). PCVV infection was lower in cells expressing high amounts of MUC1, compared with uncoated VV; however, aMUC1-PCVV resulted in restoration of infection. These results demonstrated that binding of anti-VV neutralizing antibodies can be successfully and significantly decreased by both the targeted VV (aMUC1-PCVV) and the targeted VV’s unique chemical alteration. Additionally, both PCVV and aMUC1-PCVV, compared with bare VV, demonstrated enhanced evasion of the innate immune response.
Injectable polyvinyl alcohol microgels developed by combining microfluidics technology and a Michael type addition crosslinking reaction have achieved efficient loading of OAs and cancer virus therapy138. These pH-degradable microgels extend the survival period of OA in tumor tissues and increase the accumulation of tumor OA. The simultaneous delivery of JQ1 mediated by polyvinyl alcohol microgels has also been found to significantly inhibit PD-L1 expression, thereby overcoming immunosuppression during viral therapy.
Spain-based Sagetis Biotech is working to develop an innovative polymerization technology to create secure and efficient delivery methods—a key engineering barrier in gene therapy. VIROSHIELD™ coating technology alters the behavior of OVs and improves therapeutic efficacy through multiple means. The coated Ad protects against pre-existing neutralizing antibodies by masking its surface epitopes while retaining its elevated transfection capacity. In addition, lower activation of the adaptive immune response (3-fold decrease in neutralizing antibody production) and improved blood circulation time (3-fold increase) have been observed. The company has developed Senescence-associated gene 101 (SAG101), a polymer-coated Ad currently in preclinical studies, for the treatment of pancreatic ductal adenocarcinoma139.
Application prospects
The use of OVs is limited by toxic adverse effects and neutralizing antibodies against OVs, as well as autoimmune clearance limits. A demonstrated strategy is to ensure that the drug is efficiently enriched at the lesion site, while decreasing the accumulation of the drug in normal tissues and organs. The rapid development of polymer nanocarriers has provided a viable approach to these problems. Currently, despite major advances in polymer nanocarriers, only several have been tested in preclinical animal in vivo models, and none have achieved clinical applications, primarily because of the lack of adequate means for in-depth assessment of the safety, biocompatibility, and degradability of novel polymer materials.
Although PEG has been widely used to improve the pharmacokinetics of protein drugs, increasing evidence indicates that prolonged use of PEG can cause the body to produce antibodies that considerably decrease treatment efficacy. For the polymer nanocarriers themselves, the complexity of structural design and the tediousness of material synthesis have also limited the industrial-scale production of these “smart” nanocarriers. Thus, researchers are motivated to continually develop and improve ideal biomedical polymer materials, and to explore carrier structure designs that are more amenable to clinical translation.
Albumin
Albumin is the single-chain polypeptide with the highest content in the human body (normally accounting for more than 50% of the total plasma protein content), and it comprises 585 amino acid residues. Albumin is one of the smallest proteins present in the plasma, with a molecular weight of approximately 67 kDa140. Amounts of 13 to 14 g of albumin are produced in the liver and reach the bloodstream daily. Natural recycling mechanisms transport albumin from tissues back to the circulatory space via the lymphatic system141. Moreover, albumin is a generic macromolecular carrier that facilitates the systemic circulation of a variety of endogenous substances with limited solubility, such as fatty acids and bilirubin. Additionally, albumin can bind drugs and consequently affect their biodistribution, bioactivity, and metabolism. Examples of these drugs include taxanes, sulfonamides, penicillins, and benzodiazepines142. Because of the above functions, albumin has been widely demonstrated clinically as a safe biomaterial for the design of drug delivery systems.
Advantages and disadvantages of albumin as a nanomaterial for drug delivery
Compared with other carriers, albumin as a carrier of antitumor drugs has the following advantages. 1) Excellent biocompatibility: Albumin is an endogenous substance in the human body that does not cause toxic reactions, trigger autoimmune reactions, or cause adverse reactions, such as denaturation and degradation143. 2) Unique structure and properties: Albumin’s properties endow it with stability within a certain temperature and pH range. Therefore, for most exogenous substances, albumin is an ideal carrier that can improve the stability of exogenous substances144. 3) Excellent drug loading performance: Albumins have a distinct spatial structure and can encapsulate drugs in the form of physical encapsulation or chemical bond coupling. Studies have indicated that albumin increases the solubility of hydrophobic drugs in the plasma and has excellent protective effects toward easily oxidized drugs145,146. Pharmacokinetic studies have shown that albumin avoids recognition and phagocytosis by the reticuloendothelial system, and also passively targets organs such as the liver, kidneys, and bone marrow. Furthermore, it is covalently bound to the surface of albumin. 4) Modification of various substances with targeted functions (for example, chemical modification of amino groups in active lysines on the surface of albumin)147. 5) Longer half-life in vivo: Because albumin has a negative charge in the blood, macrophages have difficulty in clearing albumin. Therefore, long-term circulation can be achieved148. Together, these advantages of albumin have laid a foundation for albumin to serve as a useful drug carrier.
However, although albumin nanomaterials have excellent biocompatibility, nontoxicity, and nonimmunogenicity, they still have shortcomings in drug delivery: 1) they have a rapid degradation rate in vivo and are easily removed from the blood circulation; 2) they may react with proteins in the blood, thus decreasing uptake of nanomaterials by tumor cells149; and 3) because of their inherent structural properties, they may be unstable in the body’s environment, which contains various enzymes and protein complexes150.
Advances in albumin-encapsulated OVs
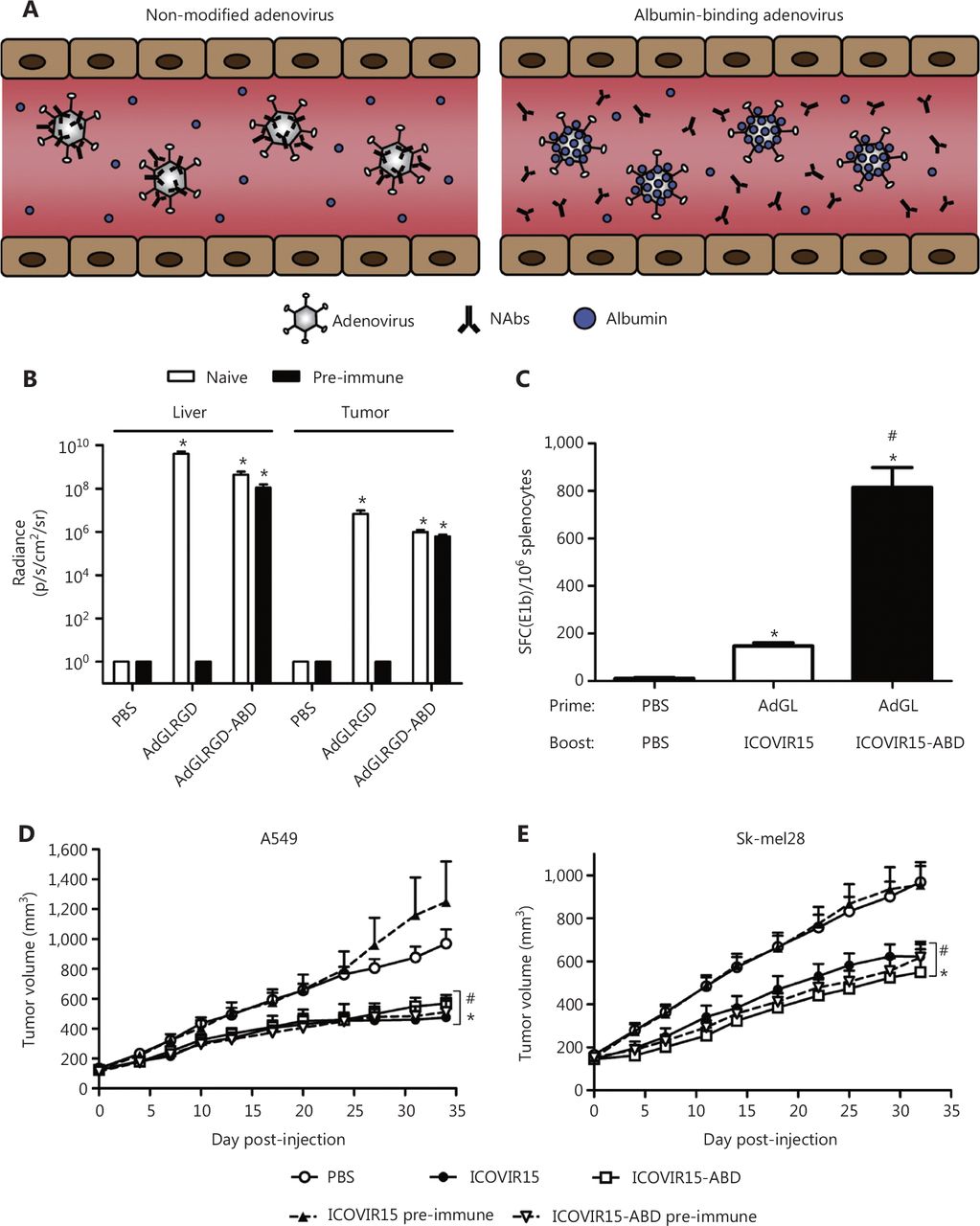
The binding of human and mouse albumin is mediated by insertion of an albumin-binding domain (ABD) into the hypervariable region 1 (HVR1) of the Ad capsid151 (Figure 7). After systemic viral treatment, albumin shields the capsid against NABs. The modified virus is totally neutralized in mice after Ad vaccination, and the ABD insertion safeguards the virus, thus enabling the same organ transduction and oncolysis as that observed in naive mice. Therefore, the ABD-mediated protection of viral capsids is an effective method to circumvent pre-existing NABs. This strategy has translational relevance in the use of adenoviruses for gene therapy, cancer virus therapy, and vaccination.

The albumin coating of the viral capsid effectively escapes pre-existing NABs. (A) Diagrammatic sketch. (B–E) ABD-modified adenoviruses evade NABs after systemic administration in pre-immune mice. Copyright © 2016 Elsevier B.V.
VCN-11, a novel Ad, has remarkable potential for eradicating of cancer cells. Ad VCN-11 was genetically modified to express hyaluronidase (PH20) and display ABD on the hexon. The virus can be coated with albumin, because of the presence of ABD, thus enabling avoidance of NABs as it enters the bloodstream. Using several tumor models, Mato-Berciano et al.152 have examined tumor targeting and anticancer activity in the presence of NABs. In tumor cells, compared with normal cells, VCN-11 showed a 450-fold greater cytotoxicity but less systemic toxicity. Thus, higher viral loads could be administered with a pre/post dosing technique, thereby improving tumor targeting. In addition, VCN-11 demonstrated efficient tumor targeting in the presence of high amounts of NABs in vivo, whereas control viruses without ABD were destroyed by NABs from various sources. Given these unique qualities, a proof-of-concept clinical trial is anticipated to analyze pre/post dosage fractionation techniques and repeat dosing, to determine the safety and tumor targeting abilities of VCN-11.
Application prospects
In recent years, albumin has received substantial attention in nanodrug delivery systems, because of its excellent biocompatibility and low toxicity. The albumin nanodelivery system is characterized by extended circulation and tumor targeting, both of which significantly improve the therapeutic effects of the drug. In the future, an attractive research direction for albumin-based OV delivery systems is the design of environment-responsive albumin therapeutic systems. Tumor tissue has unique properties from those of normal tissue, such as an acidic environment, hypoxia, highly reactive oxygen species, and enzyme overexpression, all of which are fundamental for the study of responsive albumin nanotherapeutic systems. Albumin as a drug carrier has several drawbacks, such as limited sources of human serum albumin, a mild immune reaction to bovine serum albumin used in injections, and susceptibility to denaturation. Recombinant human serum albumin, a genetically engineered protein expressed by yeast cells, has been developed in recent years and can be used as an alternative to human serum albumin. The key to the clinical application of albumin nanofabrication is to identify stable raw materials and extremely reproducible preparation methods. An increasing number of albumin and OV-based drugs are expected to achieve clinical translation in the future.
Other nanoparticles
In addition to the 3 types of nanoparticles described above, numerous biological techniques have been used for the systemic administration of OVs, including extracellular vesicles (EVs), encapsulation OVs, biomimetic mineralization of OVs, and hybrid carrier systems of OVs and organic framework materials. These biomaterials have excellent biocompatibility and in vivo targeted delivery performance, thereby broadening the range of applications for OV therapy for tumors.
Advances in systemic delivery of extracellular vehicle-encapsulated OVs
EVs are membrane vesicles that are expelled by cells, and that transport proteins, carbohydrates, RNA, and DNA, thereby aiding in intercellular communication153. EVs have several benefits, including avoiding product identification and elimination in the body, crossing physiological obstacles, and having excellent biocompatibility and stability. The ability of these EV encapsulated medications to achieve tumor tropism is enabled by EVs’ susceptibility to artificial gene modification.
Using 2 separate biomimetic synthesis techniques, Lv et al.154 have created bioengineered cell membrane nanocapsules with tailored ligands to obtain significant antiviral immune shielding and targeting capabilities for oncolytic therapy. The second method is based on clustered regularly interspaced short palindromic repeats (CRISPR) engineering to express targeted ligands in red blood cell membranes in vivo. The first method uses in vitro genetic membrane engineering to embed targeted ligands into cell membranes. The findings have demonstrated that both bioengineering techniques preserve OA’s capabilities for infection and replication in the presence of NABs, in both in vitro and in vivo settings.
Ad5D24-CpG and paclitaxel (PTX) have been encapsulated in EVs by Garofalo et al.155,156, and found to significantly improve transduction efficacy in vitro, thus enhancing infection titer and cytotoxicity, and consequently anticancer effects in lung cancer models. Imaging technology has demonstrated that Ad encapsulated with EVs intravenously exhibits tumor tropism, whereas tumor selective delivery has not been observed through the intraperitoneal administration pathway.
To enhance the efficacy of cancer virotherapy by augmenting tumor cell autophagy, Ban et al.157 have used bacterial outer membrane vesicles (OMVs) containing Ads as microbial nanocomposites. These OMVs slow internal circulation clearance of OMV surface antigen and promote tumor formation by encasing itself in a biomineral shell. The catalytic activity of overexpressed pyranose oxidase (P2O) from microbial nanocomposites enhances oxidative stress levels and initiates tumor autophagy after tumor cell entry. The autophagy-induced autophagosomes further promote Ads replication in infected tumor cells, leading to Ads-overactivated autophagy. OMV, a potent immune stimulant, has been used in preclinical cancer models in female mice to alter the immunosuppressive tumor microenvironment and stimulate the anti-tumor immune response.
Advances in biomineralized OV
OAs, because they preferentially reproduce in high titer tumor cells, provide an excellent option for clinical anticancer therapy. By encasing OAs in calcium carbonate and manganese carbonate (MnCaC) biomineral shells, Huang et al.158 have engineered an OA that prevents the immune system from clearing the virus, thereby prolonging its internal circulation. After assembling at the tumor site, MnCaCs dissolve and release Mn2+, thus resulting in conversion of endogenous H2O2 to oxygen (O2), increasing the ability of OA to replicate, and markedly increasing antitumor efficacy. A simultaneous rise in Mn2+ and O2 makes T1 mode magnetic resonance imaging and photoacoustic imaging feasible, and allows for real-time monitoring data in therapy.
Conclusions and future perspectives
Intravenous delivery of OVs is a clinical issue that must urgently be addressed. Many preclinical studies have sought to determine the optimal delivery method, but no OV drug that can currently be administered intravenously to treat tumors. On the basis of these preclinical studies, we believe that liposomes, polymers, and albumins are the most promising nanomaterials to address these issues. Because many related drugs that have successfully entered the clinic have been packaged and delivered with liposomes, polymers, and albumins, their safety and practicality have survived clinical testing.
However, several critical issues in the delivery of OVs with these 3 nanocarriers must be addressed. The tumor microenvironment is extremely complex, and the targeting and therapeutic effects of nanomaterials are extremely limited. Because endogenous signals inside and outside tumor cells are difficult to control and vary among individuals, therapeutic effects in clinical practice are often not uniform. To address these problems, nanomedicine delivery systems with multiple targeting mechanisms can be designed. Unlike a single target, nanocarriers can carry 2 or more “guidance” signals simultaneously, thus improving the probability and accuracy of identifying patient tissues. In addition, bionic in vitro test platforms and preclinical models that can efficiently evaluate the performance of nanocarriers must be developed. Given the rapid advances in nanotechnology and the accumulation of relevant experience in recent years, we believe that these 3 newcomers will play an increasingly important role in the development of OVs. We also hope that continuous improvements will be made in personalized treatment approaches for OVs based on these 3 nanocarriers, to offer new hope in the treatment of malignant tumors.
Conflicts of interest statement
No potential conflicts of interest are disclosed.
Author contributions
Conceived and designed the analysis: Liting Chen, Chen Xu, Xiao Zhao, Funan Liu.
Collected the data: Liting Chen, Chen Xu, Defang Ouyang, Shuhui Song, Youbang Xie.
Contributed data or analysis tools: Defang Ouyang, Youbang Xie, Xiao Zhao, Funan Liu.
Performed the analysis: Liting Chen, Zhijun Ma, Chen Xu.
Wrote the paper: Liting Chen, Zhijun Ma, Chen Xu, Xiao Zhao, Funan Liu.
Footnotes
↵*The authors contributed equally to this paper.
- Received July 25, 2023.
- Accepted September 25, 2023.
- Copyright: © 2023, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.
- 8.↵
- 9.↵
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.
- 114.
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.