

Abstract
Objective Cancer vaccines that rely on tumor antigen-specific CD8+ T cell responses, are promising anti-cancer adjuvant immunotherapies. This study investigated whether genetically attenuated Plasmodium sporozoite (GAS) could be used as a novel vector to induce antigen-specific CD8+ T cell responses against lung cancer.
Methods We constructed GAS/MAGE-A3, a recombinant GAS engineered to express the lung cancer-specific antigen, melanoma-associated antigen 3 (MAGE-A3), and assessed its therapeutic effects against lung cancer.
Results Robust parasite-specific CD8αlowCD11ahigh and CD49dhighCD11ahigh CD4+ T cell responses as well as a MAGE-A3-specific CD8+ T cell response were induced in GAS/MAGE-A3-immunized mice. Adoptive transfer of GAS/MAGE-A3-induced CD8+ T cells from HLA-A2 transgenic mice into lung cancer-bearing nude mice inhibited tumor growth and prolonged survival.
Conclusions These findings demonstrate that GAS/MAGE-A3 induces a strong MAGE-A3-specific CD8+ T cell response against lung cancer in vivo, and indicate that GAS is a novel and efficacious antigen delivery vector for antitumor immunotherapy.
keywords
Introduction
Lung cancer remains the most common cause of cancer-related death, resulting in 1.35 million deaths annually and accounting for ~30% of all cancer deaths in the world1. Traditional anticancer therapies, including surgery, chemotherapy, and radiotherapy, are often effective in treating early-stage lung cancer; however, most patients in advanced stages are not good candidates for traditional treatments. Despite improvements in anti-lung cancer targeted therapies, such as those targeting epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) fusion oncogenes, most patients do not achieve long-term disease control, and median overall survival rates remain low2-4. Additionally, resistance to these targeted drugs usually develops after 6–12 months of treatment5. Therefore, novel therapies are needed to improve prognoses of lung cancer patients.
Vaccines that rely on a patient’s immune system to elicit an antigen-specific CD8+ T cell response against tumor cells are promising anti-lung cancer adjuvant immunotherapies. A variety of strategies for cancer vaccine production have been developed, with the use of whole-tumor cells, DNA-bearing viral vectors, proteins, and peptides. However, current vaccines do not hitherto elicit a strong enough tumor antigen-specific CD8+ T cell response to be highly effective6.
Malaria, one of the most devastating diseases worldwide, results from infection with the parasite, Plasmodium falciparum. However, knocking out specific genes, such as “upregulated in infective sporozoites 3” (UIS3), UIS4, or Fabbf completely attenuates the malaria parasite sporozoites, resulting in a genetically attenuated sporozoites (GAS)7,8. GAS can still infect hepatocytes, but do not pass through the liver stage, are unable to generate a blood-stage infection, and subsequently cannot cause disease7. Immunization with GAS induces sterile protective immunity against malaria parasite infection and is a promising strategy for preventive malaria vaccination9-13. Our previous study showed that GAS activate innate immunity in a subcutaneous Lewis lung cancer model14. As sterile protective immunity induced by GAS is largely dependent on parasite-specific CD8+ T cell responses15,16, we hypothesized that engineered GAS could be used as vectors to induce robust anti-tumor immune responses, including tumor antigen-specific CD8+T cell and non-specific anti-tumor immune responses.
A crucial factor in designing cancer vaccines is choosing cancer-specific antigen targets that would not affect normal tissues. Melanoma-associated antigen 3 (MAGE-A3), a member of the cancer testis antigen (CTA) family, is highly expressed in non-small cell lung cancers (NSCLCs)17,18; and MAGE-A3-based anti-lung cancer immunotherapies are currently being developed19. Thus, MAGE-A3 can be considered as a candidate antigen for a vaccine against lung cancer. In this study, we generated a recombinant GAS expressing the human MAGE-A3 protein using the CRISPR-Cas9 system and investigated whether this GAS could induce robust MAGE-A3-specific CD8+ T cell responses as well as inhibit the growth of subcutaneously implanted lung tumors in nude mice.
Materials and methods
Mice, cell lines, and parasite
HLA-A2 transgenic mice [B6.Cg-Tg(HLA-A/H2-D)2Enge/J] purchased from the Jackson Laboratory (stock no: 004191; Bar Harbor, ME, USA) and nude mice purchased from the Nanjing Biomedical Research Institute (Nanjing University, Nanjing, China) were kept under standard pathogen-free conditions. Female mice (6–8 weeks old) were weight-matched for use in different experimental groups. All the animals were cared for according to the Animal Care Guidelines of the Third Military Medical University. The human lung cancer cell line, A549 (TCHu150), and HepG2 (TCHu72) liver cancer cells, were purchased from the cell library of the Chinese Academy of Sciences. A549-luciferase (A549-Luc) was purchased from Shanghai Model Organisms Center (NM-F04-1). The Plasmodium berghei (P. b.) ANKA strain was a gift from a senior investigator, Xinzhuan Su, of National Institute of Allergy and Infection Diseases (NIAID), Bethesda, MD, USA.
MAGE-A3 expression in lung cancer cells and tissues
MAGE-A3 expression in A549 cells was detected using Western blot and immunofluorescence assays. For Western blot, cells were lysed in lysis buffer; supernatant was collected by centrifugation and separated by SDS-PAGE. After transfer, polyvinylidene difluoride (PVDF) membranes were incubated with 1:1000 anti-MAGE-A3 (sc-33234, Santa Cruz Biotechnology, Dallas, TX, USA) followed by incubation with 1:4000 rabbit anti-goat HRP-conjugated secondary antibody (ZB-2306, ZSGB, Beijing, China). The membranes were developed using Western BLoT Chemiluminescence HRP Substrate (T7101Q, Takara Biomedical Technology, Beijing, China). For immunofluorescence assays, cells fixed on slides were labeled with 1:200 anti-MAGE-A3 (sc-33234, Santa Cruz Biotechnology, Dallas, TX, USA) followed by 1:100 IFKine Red AffiniPure donkey anti-goat IgG (H+L) (#A24431, Abbkine, Wuhan, China). Nuclei were counterstained with DAPI (C1005, Beyotime, Shanghai, China). Coverslips were affixed on slides using DAKO Fluorescence Mounting Medium (S3023, Agilent, California, USA). MAGE-A3 expression was detected using immunohistochemistry (IHC) in subcutaneously implanted A549 cell tumors from mice, and in tumor and tumor-adjacent tissues from NSCLC patients. In brief, after incubating with anti-MAGE-A3 and anti-mouse IgG HRP-conjugated secondary antibody, tissue sections were treated with 3, 3’-diaminobenzidine (DAB) for 2 min and scored for the extent of MAGE-A3 expression as follows: 0, 0%–5% MAGE-A3-positive cells; 1, < 25% MAGE-A3-positive cells; 2, 25%–50% MAGE-A3-positive cells; 3, 50%–75% MAGE-A3-positive cells; and 4, 75%–100% MAGE-A3-positive cells.
Recombinant parasite construction and characterization
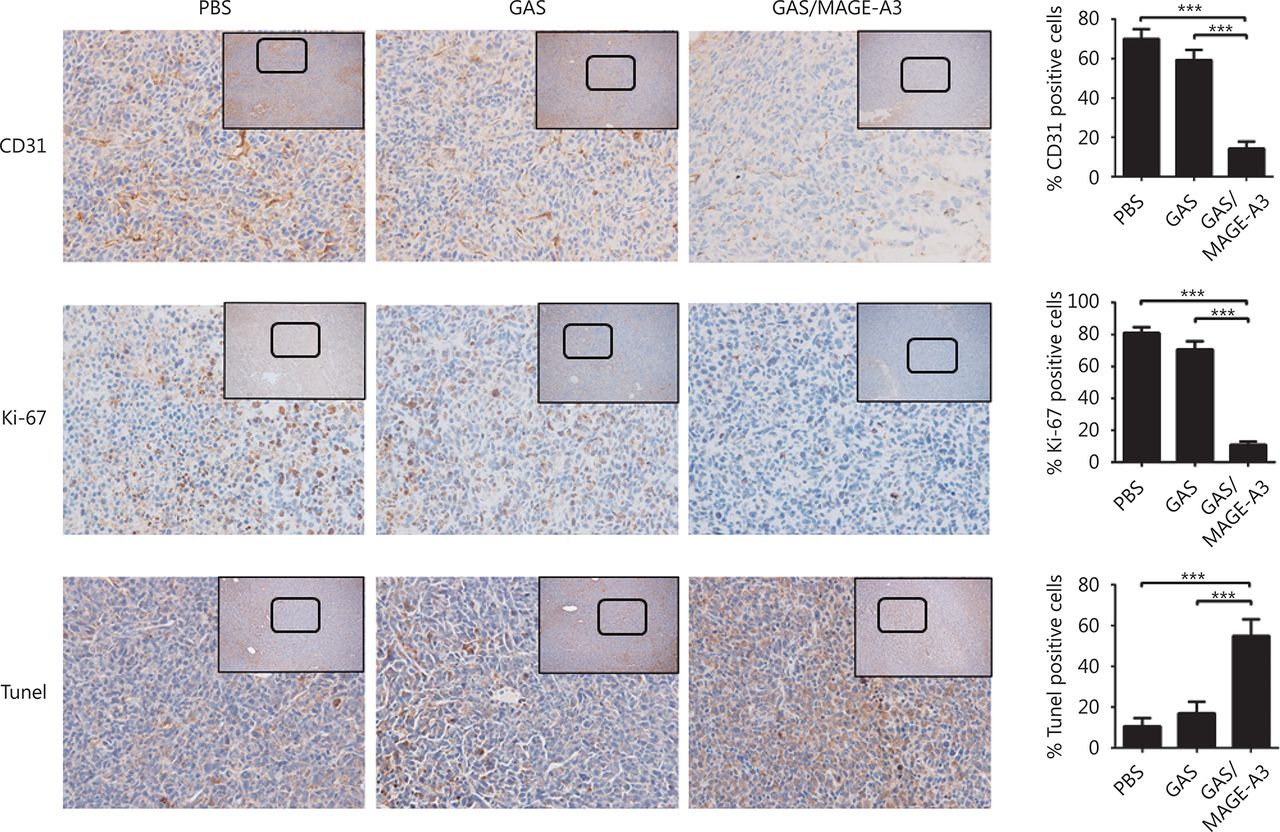
Recombinant parasites were constructed by using CRISPR-Cas9 as previously described20,21. Plasmid pYC expressing DNA endonuclease enzyme (Cas9) and pyrimethamine resistance genes (a gift from Professor Yuan Jing of Xiamen University) was used to construct a genetically attenuated, MAGE-A3-expressing sporozoite (GAS/MAGE-A3). First, sgRNA (5’-GCAATTGCTTTGTTATCATC-3’), specifically targeting P.b. ANKA UIS3 gene, was inserted downstream to the Plasmodium U6 promoter in the pYC plasmid. Second, the homologous recombinant fragment for replacing the whole UIS3 coding sequence (856 bp) containing a 5’UTR of UIS3 [coding sequence of human MAGE-A3 (900 bp)] and a 3’UTR of UIS3 was constructed by overlapping PCR and inserted into multiple clone sites of the pYC plasmid. Third, mature P.b. ANKA schizozoites were synchronized and collected as previously described22. Ten micrograms of purified recombinant plasmids containing sgRNA and a homologous recombinant fragment was electroporated into mature P.b. ANKA schizozoites by using the Amaxa human T cell Nucleofector kit (VPA-1002, Lonza, Basel, Switzerland). Fourth, parasites were immediately injected into BALB/c mice via tail vein injection, and mice were fed water with 8 μg/mL pyrimethamine (Sigma, St. Louis, USA) for 10 d, in order to screen positive parasites. Later, the pyrimethamine-resistant parasites were cloned as described (Supplementary Figure S1). The integration of the human MAGE-A3 coding sequence downstream to the UIS3 5’UTR of the cloned parasite genome was confirmed by PCR and DNA sequencing.

MAGE-A3 expression in A549 cells, subcutaneous tumors, and clinical lung cancer tissues. (A) Western blot analysis of MAGE-A3 in A549 cells (Cell) and subcutaneous tumors (Sub). (B) Immunofluorescence analysis of MAGE-A3 in A549 cells. IHC analyses of MAGE-A3 in A549 subcutaneous tumors (C) and in NSCLC patient tumors and adjacent tissues (D) (40 x). (E) Statistical analysis of MAGE-A3 expression in NSCLC patient tumors and adjacent tissues. *** P ≤ 0.001.
Female Anopheles stephensi mosquitoes were fed to GAS and GAS/MAGE-A3-infected mice kept at 20–21°C and 70%-80% relative humidity. Twenty days post-infection, the salivary glands of the mice were dissected and collected in RPMI 1640 containing 2.5 μg/mL amphotericin B, 100 U/mL penicillin, and 100 μg/mL streptomycin (Sangon Biotech, China). Sporozoites were released by grinding the salivary glands using a plastic grinding bar in a 1.5 mL Eppendorf (EP) tube, and the debris in the suspension was filtered using a 200-mesh nylon mesh. Using a blood count plate, the sporozoites in the filtered suspension were counted. HepG2 cells were infected with the fresh isolated sporozoites in ratio of 3:1 and incubated for 24 h. Because expression of MAGE-A3 is driven by the UIS3 promoter, which is only activated after the parasite has developed into a sporozoite in the salivary gland, MAGE-A3 expression by GAS/MAGE-A3 was detected 24 h after sporozoite invasion into HepG2 cells. For this experiment, HepG2 cells were labeled with 1:200 anti-MAGE-A3 antibody and 1:100 IFKine Red AffiniPure donkey anti-goat IgG (H+L). MAGE-A3 expression in GAS/MAGE-A3-infected HepG2 cells was observed under a confocal microscope (LSM780NLO; Carl Zeiss, Oberkochen, Germany). We lysed 5 × 106 GAS/MAGE-A3 sporozoites to detect MAGE-A3 using Western blot as described above for human cells.
Immunization of HLA-A2 transgenic mice
HLA-A2 transgenic mice were immunized intravenously three times at 2-week intervals with either phosphate-buffered saline (PBS), or 5 × 104 GAS, or GAS/MAGE-A3.
Flow cytometry
Single-cell mouse splenocyte suspensions were prepared 2 weeks after the final immunization. For CD49dhighCD11ahigh CD4+ T cell detection, splenocytes were incubated with anti-mouse CD4-APC/Cy7 (Biolegend, USA), anti-mouse CD49d-FITC (Biolegend, USA), and anti-mouse CD11a-PE (Biolegend, USA). To detect CD8αlowCD11ahigh T cells, cells were incubated with anti-mouse CD8α-PerCP5.5 (Biolegend, USA) and anti-mouse CD11a-PE (Biolegend, USA). Fluorescence-activated cell sorting (FACS) was performed on a FACSCalibur flow cytometer (BD Biosciences) and data were analyzed using CellQuest Pro (BD Biosciences) software.
IFN-γ-secreting CD8+ T cell detection by ELISPOT
Spleen cells (5 × 105) were collected from mice 7 d after the last immunization and 10 μg/mL of peptide (EVDPIGHLY) was added to a 96-well plate. Plates were washed, treated sequentially with biotinylated anti-mouse IFN-γ (eBioscience, USA) and streptavidin-HRP (eBioscience, USA) for 1 h, and finally developed. Plates were dried overnight and analyzed using the Champ spot III EliSpot Reader (Saizhi, China). Spot-forming cells were counted using Saizhi EliSpot Reader Software (Saizhi, China) and revised manually if it was necessary. Data were expressed as the number of spot forming cells per 1 × 105 input lymphocytes.
Adoptive transfer of CD8+ T cells from immunized HLA-A2 transgenic mice to nude mice
After final immunization, CD8+ T cells were purified from single-cell splenocyte suspensions from either GAS- or GAS/MAGE-A3-immunized HLA-A2 transgenic mice, with positive selection using a MACS cell separation system (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. Subsequently, 1 × 107 purified CD8+ T cells (> 95% purity) were injected intravenously into nude mice 10 d after the mice were subcutaneously implanted with A549 cells.
Tumor growth following adoptive transfer of CD8+ T cells
A549 or A549-Luc cells (5 × 106) in 200 μL of 0.9% saline were injected subcutaneously into nude mice to generate a human tumor-xenograft animal model. CD8+ T cells (1 × 107) purified from either GAS or GAS/MAGE-A3 HLA-A2 transgenic mice were intravenously injected into nude mice 10 d after A549 cell implantation. Tumor sizes were measured three times weekly using the following formula: tumor volume = length × width × width/2. At 10 and 42 d, mice were anesthetized and intraperitoneally injected with 150 mg/body weight (g) of D-luciferin (122796, Perkin Elmer, Waltham, MA, USA). Bioluminescence imaging was used to monitor the tumor growth.
Tumor cell proliferation, apoptosis, and angiogenesis
Tumors were resected for histopathological examination 32 d after adoptive transfer of CD8+ T cells into tumor-bearing nude mice. Tumor tissue sections (4–5 μm thick) were fixed with 10% formalin and stained using anti-CD31 (ab32457, Abcam, Cambridge, UK) and anti-Ki67 (ab15580, Abcam) antibodies, and a terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay was also performed (G7360, DeadEnd™ Colorimetric TUNEL System, Promega, Madison, WI, USA). Sections were washed three times, treated with DAB for color development, and examined. Photographs were captured using a NanoZoomer 2.0-RS optical microscope (Hamamatsu Photonics, Hamamatsu, Japan).
Statistical analysis
For parametric data in each experiment, the difference between two groups was statistically analyzed using one-way analysis of variance (ANOVA). Survival curves were analyzed using a log-rank test. Data are presented as means ± standard deviations from three independent experiments. All statistical analyses were performed using GraphPad Prism software 6.0 (GraphPad Software, San Diego, CA, USA).
Results
MAGE-A3 protein is highly expressed in A549 cells and lung cancer tissues
To determine whether MAGE-A3 is a specific antigen related to lung cancer, we detected MAGE-A3 in both A549 cells and lung cancer tissues. Western blot demonstrated that MAGE-A3 was expressed in both the cultured A549 cells as well as in subcutaneously implanted tumor tissues (Figure 1A). Immunofluorescence assays showed that MAGE-A3 was primarily localized in the cytoplasm of A549 cells (Figure 1B). This was confirmed by immunohistochemical (IHC) analysis of A549 subcutaneously implanted tumors (Figure 1C). IHC analysis of NSCLC and adjacent tissue samples from 50 patients (Figure 1D) showed that 59% of tumor tissues were MAGE-A3-positive; however, only 5% of adjacent tissues were positive for MAGE-A3 (Figure 1E). These findings strongly suggest that MAGE-A3 might be a potential immunodominant antigen in NSCLC.
Construction and characterization of recombinant GAS/MAGE-A3
To investigate the efficacy of GAS as a vector for MAGE-A3 delivery, we constructed a recombinant GAS expressing MAGE-A3 (GAS/MAGE-A3). We replaced the Plasmodium gene, UIS3, with human MAGE-A3 coding sequence using the CRISPR-Cas9 system (Figure 2A). After electroporation, pyrimethamine screening, and cloning (Figure 2B), PCR analysis identified seven positive clones (Figure 2C) employing DNA sequencing, out of which one was verified as a recombinant, GAS/MAGE-A3 (Figure 2D). UIS3 is selectively expressed in the Plasmodium sporozoites and at exo-erythrocytic stages23; MAGE-A3 expression was driven by the UIS3 promoter in this study. Immunofluorescence and Western blot analyses performed 24 h after GAS/MAGE-A3 sporozoite invasion into HepG2 cells showed that MAGE-A3 was successfully expressed in GAS/MAGE-A3-infected cells, but not in GAS-infected cells (Figure 2E and 2F). These findings indicate that GAS could successfully deliver the lung-cancer antigen, MAGE-A3, in HepG2 cells.

GAS/MAGE-A3 construction and MAGE-A3 expression in GAS/MAGE-A3-infected HepG2 cells. (A) UIS3 replacement with MAGE-A3 coding sequence via CRISPR-Cas9. (B) Parasitemia curve for mice infected with PYC-sgRNA (UIS3)-MAGE-A3-electropated P.b. ANKA. (C) Positive clones with UIS3 successfully replaced with MAGE-A3 were identified via PCR. (D) One clone with UIS3 successfully replaced was verified via DNA sequencing. (E) At 24 h after GAS/MAGE-A3 sporozoite invasion into HepG2 cells, MAGE-A3 expression was detected in infected cells via immunofluorescence. (F) Western blot analysis of MAGE-A3 in GAS/MAGE-A3-infected HepG2 cells. Experiments were performed three times, and one is presented.
GAS/MAGE-A3 triggers a robust and specific CD8+ T cell response in HLA-A2 transgenic mice
Next, we investigated whether GAS/MAGE-A3 could induce a robust cellular immune response against malarial parasites and MAGE-A3. CD8alowCD11ahigh T cells and CD49dhighCD11ahigh CD4+ T cells, representing parasite-specific T cells against malarial infection, were detected to evaluate the immune effect of GAS/MAGE-A3 vaccine24,25. Seven days after the final immunization, we compared CD8αlowCD11ahigh and CD49dhighCD11ahigh CD4+ T cell counts, representing a parasite-specific CD4/CD8+ T cell response, in blood and spleen samples from control, GAS/MAGE-A3-, or GAS-immunized HLA-A2 transgenic mice (Figure 3A). CD49dhighCD11ahigh CD4+ T cell counts in GAS/MAGE-A3- or GAS-immunized mice blood and spleen samples were 2-fold higher than those of control mice (Figure 3B). Additionally, CD8αlowCD11ahigh T cell counts in GAS/MAGE-A3- or GAS-immunized mouse blood and spleen samples were > 4-fold higher than those of control mice ( Figure 3C). There was no difference in the ability of CD8+ T cell induction between GAS and GAS/MAGE-A3. These results suggest that integration of exogenous MAGE-A3 into GAS did not influence the GAS-induced CD8+ T cell response.
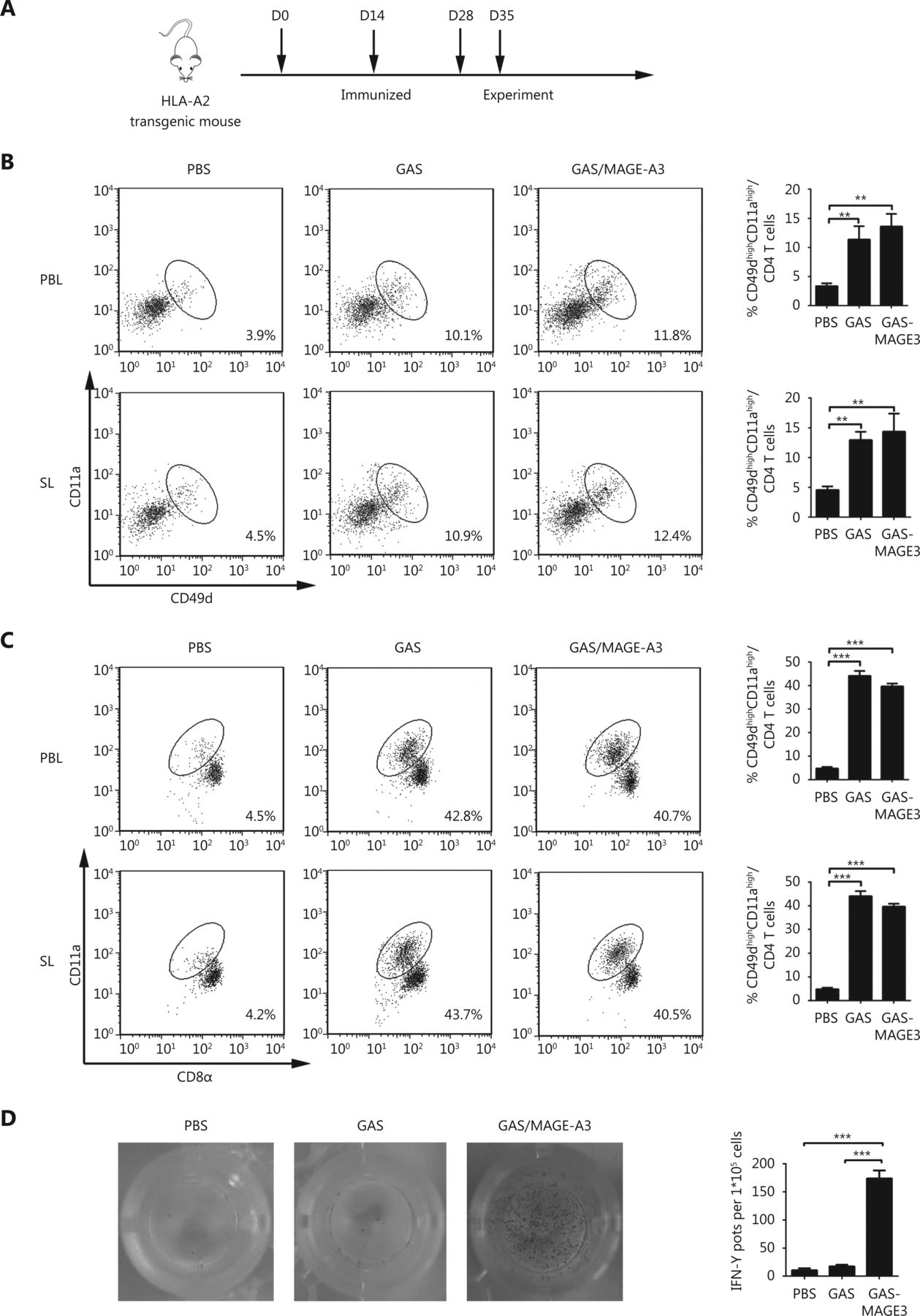
CD49dhighCD11ahigh CD4+ and CD8αlowCD11ahigh T cells in peripheral blood and spleen from immunized HLA-A2 transgenic mice. (A) Vaccination schedule and detection time points for CD49dhighCD11αhigh CD4+ and CD8αlowCD11ahigh T cells. (B) CD49dhighCD11ahigh CD4+ and (C) CD8αlowCD11ahigh T cell frequencies in peripheral blood lymphocytes (PBL) and spleen lymphocytes (SL) were assessed via FACS. (D) IFN-γ secretion by MAGE-A3-specific CD8+ T cells was detected using ELISPOT. * P ≤ 0.05, *** P ≤ 0.001.
A previous study has confirmed that CD8+ T cells are the main anti-tumor effector cells26. We therefore determined whether GAS/MAGE-A3 infection could also induce a MAGE-A3-specific CD8+ T cell response. Our ELISPOT assay results revealed that GAS/MAGE-A3 infection provoked strong MAGE-A3-specific CD8+ T cell responses. IFN-γ secreted by CD8+ T cells from GAS/MAGE-A3-immunized HLA-A2 transgenic mice was more than 15-fold higher than that by cells from control mice. In contrast, we detected no IFN-γ expression in CD8+ T cells from GAS-infected mice (P ≤ 0.001; Figure 3D). These findings suggest that GAS/MAGE-A3 could not only induce a strong cellular immune response against malaria parasites, but also could act as a vector to trigger robust specific CD8+ T cell responses against its delivered antigen, MAGE-A3.
Adoptive immunotherapy using CD8+ T cells suppresses subcutaneous A549 cell tumor growth in nude mice
To assess the therapeutic effects of MAGE-A3-specific CD8+ T cells in a mouse model, CD8+ T cells purified from GAS/MAGE-A3- or GAS-infected HLA-A2 transgenic mice spleens were adoptively transferred into nude mice 10 d after these mice were subcutaneously implanted with A549 cells (Figure 4A). At d 42 post-implantation, nude mice that received CD8+ T cells from GAS/MAGE-A3-immunized mice had smaller tumors than control mice and those that received CD8+ T cells from GAS-immunized mice (Figure 4B and 4C). Similarly, tumor volumes were reduced in nude mice that received CD8+ T cells from GAS/MAGE-A3-immunized mice (P < 0.05 vs. mice that received CD8+ T cells from GAS-immunized mice; P ≤ 0.001 vs. controls; Figure 4D and 4E). Mice that received CD8+ T cells from GAS/MAGE-A3-immunized mice also exhibited prolonged survival (Figure 4F). Tumor growth was slightly suppressed in nude mice that received CD8+ T cells from GAS-immunized mice, but survival was not improved compared to controls (Figure 4E and 4F). These findings indicate that GAS/MAGE-A3-induced MAGE-A3-specific CD8+ T cells attenuated A549 cell tumor growth in vivo.

Therapeutic effects of CD8+ T cells from immunized mice on subcutaneously inoculated A549 cell tumors in nude mice. (A) Inoculation schedule for nude mice and adoptive transfer of CD8+ T cells from immunized HLA-A2 transgenic mice. (B) Representative image and (C) statistical analysis of luminescence intensity in tumor-bearing nude mice who received CD8+ T cells purified from control, GAS-, or GAS/MAGE-A3-immunized HLA-A2 transgenic mice. (D) Representative images and (E) statistical analysis (size) of tumors removed from nude mice 32 d after adoptive transfer of CD8+ T cells from control, GAS-, or GAS/MAGE-A3-immunized HLA-A2 transgenic mice. (F) Survival curves for tumor-bearing nude mice treated with CD8+ T cells from control, GAS-, or GAS/MAGE-A3-immunized HLA-A2 transgenic mice. ** P ≤ 0.01.
Effects of CD8+ T cell adoptive immunotherapy on tumor cell proliferation, apoptosis, and angiogenesis
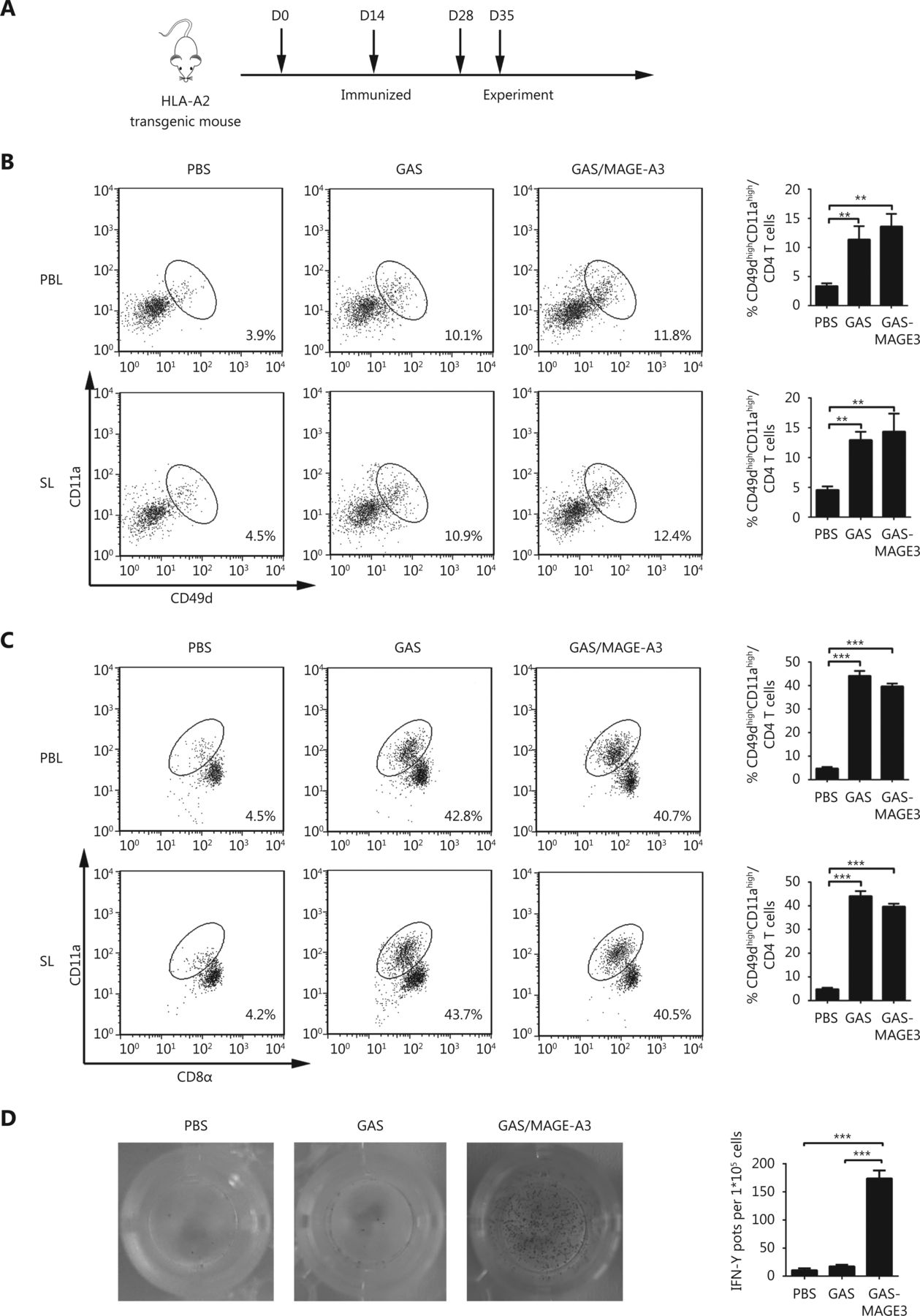
A TUNEL assay and IHC analysis of the proliferation marker, Ki-67, and angiogenesis maker, CD31, were performed using tumor tissues from nude mouse 42 d after adoptive transfer of CD8+ T cells from GAS- or GAS/MAGE-A3-immunized mice. The TUNEL assay results showed increased numbers of apoptotic cells in tumors from mice that received CD8+ T cells from GAS/MAGE-A3-immunized mice. Furthermore, only 10%-15% CD31 (blood vessel endothelial cells marker) and Ki-67 (proliferation marker) positive cells were found in tumors from mice treated with adoptive CD8+T cells, these results indicated that CD8+ T cells induced by GAS/MAGE-A3 significantly reduced tumor cell proliferation and decreased tumor vasculature (Figure 5).

Effects of adoptive immunotherapy using CD8+ T cells on tumor cell proliferation, apoptosis, and angiogenesis. At 32 d after adoptive transfer of CD8+ T cells from control, GAS-, or GAS/MAGE-A3-immunized HLA-A2 transgenic mice into tumor-bearing nude mice, tumors were removed. CD31 and Ki-67 were analyzed via IHC (40 x), and tumor cell apoptosis was analyzed using TUNEL. Representative image (left), and statistical analysis (right) of CD31 and Ki-67 expression and tumor cell apoptosis. Bar graphs show mean percentage ± standard deviation. *** P ≤ 0.001. Bars: 100 μm.
Discussion
Using the CRISPR-Cas9 system, we successfully engineered GAS to express human MAGE-A3. Adoptive transfer of CD8+ T cells from GAS/MAGE-3-immunized HLA-A2 mice prolonged survival in nude mice bearing subcutaneously implanted A549 cell tumors. Our findings show that the engineered GAS is an efficacious antigen- delivery vector for antitumor immunotherapy and induces a robust tumor antigen-specific CD8+ T cell response.
Tumor-specific CD8+ T cells play crucial roles in antitumor immunotherapy. Recent research has focused on recovering exhausted tumor-specific CD8+ T cells and enhancing the CD8+ T cell response to better recognize and target tumor cells. Immune checkpoint-blocking drugs, such as anti-programmed cell death protein-1 (anti-PD-1)/PD-ligand-1 and anti-cytotoxic T lymphocyte-associated protein 4 (anti-CTL4) can promote antitumor immunity and sustained cancer regression by inhibiting effector CD8+ T cell exhaustion27. Chimeric antigen-receptor T cell (CART) immunotherapy relies on engineered CD8+ T cells to recognize tumor cells through non-major histocompatibility complex/T cell receptor patterns28. In contrast, vaccines aim to induce a tumor antigen-specific CD8+ T cell response. Consequently, anti-PD/PD-L1 or CART combined with a tumor vaccine could greatly improve immunotherapy effects.
An appropriate antigen delivery vector is essential to induce effective antitumor immune responses, especially to generate tumor-specific CD8+ T cells. While attenuated viruses and bacteria have been studied as tumor vaccine vectors, such antigen-delivery vectors have thus far been unable to elicit a strong CD8+ T cell response29. Although CD8+ T cell response seems easy to be induced by DNA- and viral vector-based vaccines in mice, the response in humans is not as effective as in mice30. Furthermore, DNA vaccines may express oncogenes that could potentially integrate into host genomes31. However, Plasmodium can induce a strong immune response in malaria patients, and immunization with attenuated Plasmodium sporozoites could induce complete protective immunity against infection by malaria sporozoites in humans and mice32-34. This protective immunity is mainly dependent on efficient induction of CD8+ T cells15,35. Therefore, recombinant Plasmodium sporozoites could be considered as potential antigen-delivery vectors of tumor vaccines.
Recently, Trypanosoma cruzi and blood-stage malarial parasites have also been assessed as vectors to elicit robust CD8+ T cell responses against tumors36,37. Interestingly, sporozoites of malaria parasites induces innate immune responses against tumors38, and Toxoplasma gondii evokes a tumor antigen-specific CD8+ T cell response39-41. Thus, we hypothesized that engineered parasites may induce both an innate immune response and adaptive immunity against a tumor-specific antigen. In this study, we demonstrated that recombinant GAS/MAGE-A3 induced a robust MAGE-A3-specific CD8+ T cell response in a mouse model. While we did not directly investigate whether GAS/MAGE-A3 also induced non-specific anti-tumor immune responses, our previous results suggest that such responses are likely14. Additionally, evidence suggests that the sporozoite antigens may persist for more than eight weeks after immunization42. Thus, GAS has obvious advantages as a vector to induce tumor antigen-specific CD8+ T cell responses.
In addition to the effective induction of a robust anti-tumor immune response, safety is another important concern in tumor vaccine design and production. We successfully replaced the UIS3 coding sequence with that of MAGE-A3 using the CRISPR-Cas9 system, which was recently used to modify the malaria parasite genome20,43. MAGE-A3 insertion did not affect sporozoite attenuation, and blood stage parasites did not develop in mice infected with either GAS or GAS/MAGE-A3 (data not shown). The tumor antigen also did not influence induction of sporozoite-specific T cell responses, and robust CD8αlowCD11ahigh and CD49dhighCD11ahigh CD4+ T cell responses, representative of parasite-specific T cell responses, were induced in both GAS and GAS/MAGE-A3-immunized mice. It was reported that infection with blood stage tumor antigen GPC3-transgenic Plasmodium yoelii also induced GPC3-specific CD8+ T cell responses37. However, while the engineered P. yoelii was non-lethal and was cleared in 20 d, clinical symptoms were observed and safety was a concern37. On the contrary, infection by the blood stage malaria parasite often induces strong CD4+ but not CD8+ T cell responses44.
This study aimed to explore the possibility of Plasmodium sporozoites as a vector for developing a vaccine against tumors. Although the GAS/MAGE-A3 can effectively induce MAGE-A3-specific CD8+ T cells response, and retard the growth of A549 cells in mice, mass production of Plasmodium sporozoites is challenging due to its unique life cycle. Other human parasites, Toxoplasma gondii and Trypanosoma cruzi, more accessible than Plasmodium sporozoites, might be the more appropriate vectors for developing tumor vaccines.
In conclusion, our work suggests that GAS is a safe vector that induces a strong tumor antigen-specific CD8+ T cell response, and may improve outcomes in lung cancer patients.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China for Dai (Grant No. 81472188) and Zheng (Grant No. 81702247), and the Youth Development Project of TMMU for Zheng (Grant No. 2016XPY17).
Conflict of interest statement
No potential conflicts of interest are disclosed.
Supplementary materials

Construction of recombinant GAS/MAGE-A3 via CRISPR-Cas9. Blood stage P.b. ANKA parasites were collected and synchronized by culturing overnight in Roswell Park Memorial Medium-1640 with 1% penicillin-streptomycin, and then merozoites were collected via density gradient centrifugation A. Schizozoites were electroporated with endo-free PYC-sgRNA(UIS3)-MAGE-A3 plasmid using the Amaxa human T cell Nucleofector Kit B. Parasites were then injected into the tail veins of BALB/c mice, which were treated with pyrimethamine as soon as parasites appeared in blood. Parasites that re-appeared in mice after pyrimethamine treatment were collected and cloned by infecting each BALB/c mouse with a single parasite. Each parasite clone was identified using PCR and DNA sequencing.
Footnotes
↵*These authors contributed equally to this work
- Received August 30, 2018.
- Accepted December 12, 2018.
- Copyright: © 2019, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.