

Abstract
Master developmental pathways, such as Notch, Wnt, and Hedgehog, are signaling systems that control proliferation, cell death, motility, migration, and stemness. These systems are not only commonly activated in many solid tumors, where they drive or contribute to cancer initiation, but also in primary and metastatic tumor development. The reactivation of developmental pathways in cancer stroma favors the development of cancer stem cells and allows their maintenance, indicating these signaling pathways as particularly attractive targets for efficient anticancer therapies, especially in advanced primary tumors and metastatic cancers. Metastasis is the worst feature of cancer development. This feature results from a cascade of events emerging from the hijacking of epithelial-mesenchymal transition, angiogenesis, migration, and invasion by transforming cells and is associated with poor survival, drug resistance, and tumor relapse. In the present review, we summarize and discuss experimental data suggesting pivotal roles for developmental pathways in cancer development and metastasis, considering the therapeutic potential. Emerging targeted antimetastatic therapies based on Notch, Wnt, and Hedgehog pathways are also discussed.
keywords
Introduction
The development of metastasis from a primary tumor site, including epithelial-mesenchymal transition (EMT), tumor neoangiogenesis, and spread of malignancy, is a multistep phenomenon for targeting tissues and organs. The spread of malignancy results from malignant cell transport through blood vessels to target tissues and organs, the invasion of the latter by infiltrating malignant cells, and the development of secondary tumors1,2. Master developmental pathways, such as Notch, Wnt, and Hedgehog, are signaling pathways that play pivotal roles along embryonic development. The role of Notch in solid tumor and hematological malignancy initiation and development has been extensively documented. In most cases, Notch is a major oncogene associated with tumor progression to metastasis, anoikis resistance, EMT, neoangiogenesis, malignant cell proliferation, and changes in tissue microenvironment promoting the homing of metastasis-promoting cells; nonetheless, Notch may also act as a tumor suppressor3–6.
The overexpression of Wnt signaling is common in many hematological malignancies and solid tumors. Clinical and experimental evidence suggests that Wnt/β-catenin activation is critical for cancer development, angiogenesis, migration, and invasion5–7. The antagonists of Wnt pathway, such as Wnt inhibitory factor 1 (WIF-1), Dickkopf proteins (Dkks), the secreted frizzled-related proteins (sFRPs), and Disheveled-axin domain containing 1 (DIXDC1), enhance the tumorigenic and metastatic processes of various cancer typesin vitro and in vivo8–10. Similar to Notch and Wnt pathways, Hedgehog is an evolutionary and developmental pathway involved not only in cellular differentiation but also in physiological and tumorigenic control of postnatal cellular events, such as proliferation, cell death, motility, migration, and invasion5,6.
Emerging therapies targeting master developmental pathways represent promising approaches for metastasis prevention and cancer stem cell elimination (Figure 1). In this study, we summarize the data supporting a role for developmental pathways in pro-metastatic processes, emphasizing on Notch, Wnt, and Sonic Hedgehog (Shh), considering their therapeutic potential. Emerging antimetastatic strategies targeting these developmental pathways are also discussed.
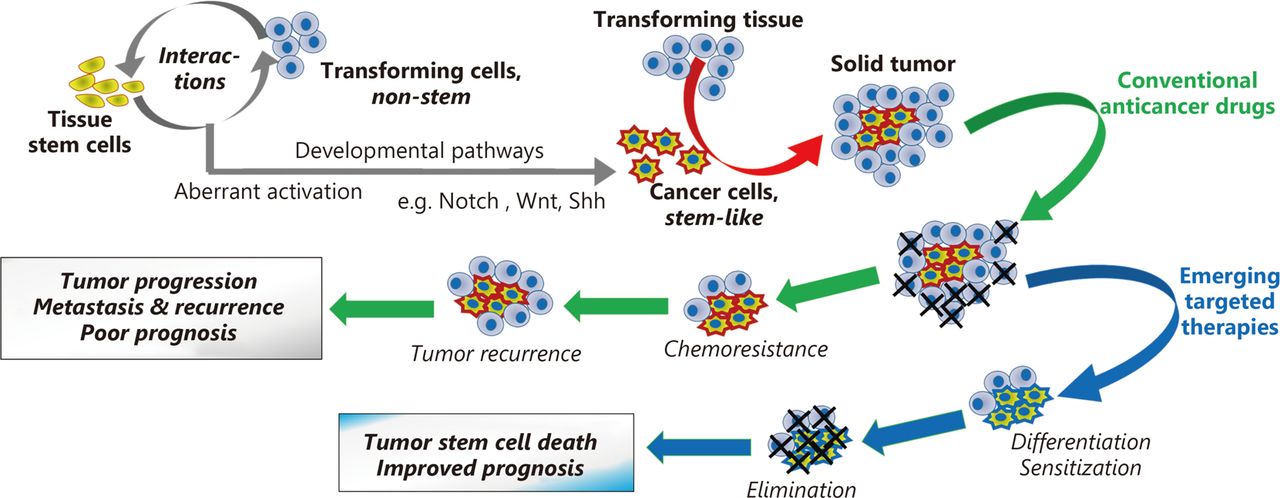
Effects of conventional anticancer drugs and of their combination with emerging therapies targeting developmental pathways on cancer stem-like cells and treatment outcome.
Pro-metastatic changes and developmental pathways
Resistance to anoikis
Similar to other anchorage-dependent cells, the survival of tumor cells, following the detachment from surrounding epithelial tissue or extracellular matrix, requires the capability to resist against the form of programmed cell death resulting from such detachment, namely, anoikis. Unlike most normal anchorage-dependent cells that undergo cell death when detached from the extracellular matrix, tumor cells can implement several molecular mechanisms, promoting their survival in suspension and the spread of metastases. The experimental and clinical evidence strongly suggests that the subversion of the repertoire of integrin developmental pathways is the main molecular mechanism favoring anoikis resistance in tumor cells.
Cell-cell and cell-extracellular matrix contacts support anchorage-dependent cell survival in an integrin-dependent manner, partly because of integrin-mediated activation of anti-apoptotic signaling pathways, such as phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), mitogen-activated protein kinase (MAPK), and focal adhesion kinase pathways11,12. The aberrant constitutive activation of these anti-apoptotic signaling pathways results in post-translational modifications and changes in the levels of Bcl-2 anti-apoptotic protein family members, i.e., multi-domain and BH3 domain-only proteins. Consequently, the overexpression of the latter proteins in tumor cells prevents the mitochondrial function disruptions that normally lead to programmed cell death13 and related pro-apoptotic mechanisms, such as metabolism-induced autophagy11,12.
In addition to these intrinsic apoptotic mechanisms, metastatic competent cells also achieve resistance to anoikis and anticancer therapy by interfering with extrinsic apoptosis pathways, such as the families of death receptors, including Fas receptors13. Notably, the prevention of Fas receptor activity results in increased levels of cellular FADD-like IL-1β-converting enzyme-inhibitory protein, a master anti-apoptotic regulator, in these tumor cells. This increase results in a cross talk between extrinsic and intrinsic caspase pathways that impair the capability to use common apoptotic caspases, particularly effector caspases, favoring tumor cell survival 13. Integrin signaling has the capability to induce ligand-independent activation of developmental signaling molecules that are able to trigger resistance to anoikis. Examples include the loss in cell adhesion factors, such as E-cadherin; and the overactivation of other EMT signaling molecules, such as the mammalian target of rapamycin (mTORC1 and mTORC2)14–16, growth factors, such as epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF) receptor signaling pathways12–15, and oncogenes, such as Wnt, β-catenin, Notch, and Shh17–20.
The potential of the pharmacological targeting of developmental signaling pathways favoring resistance to anoikis supports the survival of metastatic cells throughout the metastatic journey, i.e., from the primary site to the target tissue invasion and secondary tumor development12–16.
EMT
EMT is a reversible specialized process that occurs during the normal embryonic development, which is pivotal for cell specialization and tissue patterning. During development, an early detachment of embryonic epithelial cell from the initial tissue occurs without activation of anoikis. Then, these cells acquire the motile shape that will allow them to migrate to new sites, where they will undergo mesenchymal-to-epithelial transition, providing these cells with the capability to build new tissues21,22. This process, which also occurs during wound healing, is used by transforming cells during tumorigenesis and tumor progression. Primary tumor cells undergo EMT during cancer progression, which confers them with a mesenchymal-like phenotype associated with not only high motility but also resistance to anoikis and anticancer drugs22–24.
While normal development of EMT is controlled and highly organized, this event is more stochastic in cancer25. Physiological EMT results from the coordination of molecular events with new cell capacity. Specifically, EMT starts by losing the cell junction proteins, such as E-cadherin, claudins, occludins, and catenins, associated with the epithelial organization, followed closely by the expression of mesenchymal markers, such as N-cadherin and vimentin26,27. A large number of oncogenic factors known to induce and/or to sustain tumor cell motility and metastasis, such as transforming growth factor beta (TGF-β) and insulin-like growth factor 1, induce the loss of the epithelial marker E-cadherin28,29. E-cadherin is at the center of EMT and anoikis signaling networks because its loss is common in cancer progression, where this marker is associated with aberrant EMT, tumor cell migration and invasion, and resistance to anoikis. Moreover, the loss of E-cadherin and other adheren junction molecules disrupts epithelial tissue, leading cells to the adoption of a minimally polarized phenotype with high invasive properties. The switch in cell morphologies and properties is mediated by key transcription factors, namely, EMT-activating transcription factors. Examples include the transcription factors of a snail family (e.g., snail, slug, and smuc), zinc-finger E-box-binding (ZEB) factors [e.g., the two-handed zinc-finger factors of d-crystallin/E2 box factor (dEF1) family proteins dEF1/ZEB homeobox 1 and Smad-interacting protein 1/ZEB2], and basic helix-loop-helix factors (e.g., Twist1, Twist2, E12, E47, and Tbx3)30–32.
Motility and migration
The metastatic competent cells detached from primary tumors require being migrated to invade other sites. The migration and other motility events result from specific cytoskeletal changes mediated and regulated mainly by small GTPase proteins of the Rho family33–36. The members of the Rho GTPase family process extracellular signal into information that will lead to the reorganization of the cytoskeleton. As discussed above, the molecular events that trigger EMT and confer anoikis can activate the migratory machine in tumor cells through the members of the Rho GTPase family35.
Mounting evidence supports Rho GTPase involvement in all tumorigenic events from the primary tumor initiation to the metastasis. These small GTPases direct actin dynamic and protrusive patterns in migrating cells, including the projection of the cytoskeletal protein actin on the leading edge of the cell (lamellipodia), the extension of slender cytoplasmic projections beyond the leading edge of lamellipodia (filopodia), and the extracellular matrix degradation-associated protrusions of the plasma membrane (invadopodia)36. Actin dynamic results from its polymerization induced by actin-related protein 2 (Arp2) and Arp3. Invadopodia is a characteristic of invasive tumor cells, whereas the other patterns can occur in normal processes37–40. Rho-A, Cdc42, and Rac1 are the most documented Rho GTPases involved in cell migration and metastasis. Besides these common properties, Rho-A is also linked to stress fiber, whereas Cdc42 and rac1 are involved in filopodia and lamellipodia, respectively37,38.
In addition to the EMT-induced protrusive processes, migrating tumor cells can undergo collective amoeboid transition and mesenchymal to amoeboid transition with characteristic invadopodia39,40. Notably, such invadopodia require the enzymatic digestion of the extracellular matrix by the leading edge, mediated by matrix metalloproteinases. This type of migration is observed in many solid tumors, including breast carcinoma41.
Neoangiogenesis
The angiogenic processes triggered during tumorigenesis, namely, neoangiogenesis, provide the neoplastic tissue with new blood vessels that will supply the high requirements of this tissue in nutrients and oxygen. Such high requirements are triggered by paracrine growth signaling aberrantly activated directly or indirectly in transforming cells in response to extracellular matrix digestion41,42. The new blood vessels are also used as highways by invading tumor cells, which are able to travel great distances in the body to start metastases in other organs because of the motility provided by EMT.
Neoangiogenic dynamic relies on angiogenic signaling molecules of the VEGF family (VEGF-A/B/C/D) and their receptors (VEGFR-1/2/3/4)43,44. The main activator of the angiogenic cascade in the transforming and neoplastic tissue is the hypoxic microenvironment. The low oxygen tension observed in the microenvironment induces the stabilization of hypoxia-inducible factors (HIFs), preventing the von Hippel-Lindau binding protein-mediated degradation of these transcription factors. Then, the HIFs enter the tumor cell nuclei, where they dimerize and associate with p/300 to form a transactivation complex. Consequently, this transcriptional complex will stimulate the expression of hypoxia target genes by binding to their hypoxia response elements. Hypoxia target genes include genes involved in the angiogenesis cascade, such as VEGF system genes, and other ligand-receptor systems with high angiogenic potential, such as angiopoetin/Tie2, platelet-derived growth factor (PDGF)/PDGF receptor, Delta-like ligand 4 (DLL4)/Notch, and fibroblast growth factor (FGF)/FGF receptor45–47.
Notch signaling and metastasis
Notch family of signaling molecules
The Notch family of the transmembrane proteins encompasses four receptors (Notch1-4) and five ligands (Jagged1/2, Dll-1/3/4). The Notch receptors are activated when they bind to a ligand expressed on the membrane of an adjacent cell. Then, the receptors undergo two proteolytic cleavages at S2 and S3 sites performed by ADAM10/17 metalloprotease and gamma secretase complex, respectively. The two proteolytic events result in the release of an intracellular active form of the receptors, namely, Notch intracellular domain (NICD). Subsequently, the NICD domains enter the nucleus, where they form a transcriptional activation complex with MALM1 and RBP-jk proteins. This complex will promote the expression of various genes involved in cell fate determination, including myc, cyclinD, and genes of the helix basic family, such as hes1 and hey1. Functionally, the products of this transcription will control the cell proliferation, cell death, adhesion, invasion, and migration.
Notch in the metastatic cascade
The survival pathways of Notch signaling controls are important for anoikis resistance, such as NF-κB, Akt, Sat3, and mTOR31. The capability to modulate the expression of the EMT transcription factors, such as snail, slug, ZEB1/2, TGF-β, FGF, and PDGF, increases the expression of the mesenchymal markers (e.g., slug), downregulates the epithelial markers, and activates the Rho GTPases3,18,48. For example, Kwon et al.7 assessed the Notch signaling's pro-metastatic role in a mouse model of prostate cancer. The constitutive expression of the Notch1 intracellular domain in mouse prostate luminal cells (i) impaired secretory functions; (ii) suppressed anoikis via hes1 non-dependent NF-κB activity; and (iii) stimulated luminal cell proliferation by potentiating PI3K/Akt signaling. Recently, Shao and collaborators38 emphasized the Jagged1-induced Notch signaling triggered migration, invasion, and a slug-dependent EMT transition in breast cancer cells. All these effects were abrogated by Notch silencing48, underlining the potential of Notch targeting for the modulation of tumor cell motility. Furthermore, Rho GTPase upregulation is common in patients diagnosed with T-cell acute lymphoblastic leukemia (T-ALL), a hematological malignancy, where Notch activating mutations are observed in more than 50% of the cases49.
Notch targeting as antimetastatic strategy
Notch-based therapeutic strategies
Notch-based therapeutic strategies were developed for various steps of the signaling cascade, including ligand-receptor interactions, enzymatic cleavages of the receptors, cytoplasmic interactions, and transcriptional activation complex. The requirement of S2 and S3 cleavages for the activation of Notch receptors made these events particularly attractive for the modulation of Notch-induced repression of E-cadherin, which associates with a number of pro-tumorigenic events and poor prognosis in solid cancers, such as breast cancer50. S2 and S3 are due to ADAM10/17 metalinebreak and secretase complex, respectively. Unlike S2 cleavage inhibitors, gamma secretase inhibitors (GSI) have been widely used in anticancer preclinical research, partly due to GSIs were developed first and tested in Alzheimer’s disease and experimental models, where they proved relatively safe51. GSIs achieving a total blockade of Notch signaling display strong antineoplastic responses, while even those achieving partial blockade can exhibit decent antineoplastic activities52,53.
GSI treatment mitigated the development of tumor cell invasion and metastasis in vivo and in vitro by preventing EMT, migration, invasion, and neoangiogenesis54. A number of GSIs recently entered clinical trials, including BMS906024, MK0752, PF03084014, and R0492909755. PF03084014 is currently in phase 1 trials in metastatic pancreatic adenocarcinoma patients not previously treated with anticancer therapies. R04929097 is in phase 2 trials in metastatic melanoma56. MK0752 is in phases 1 and 2 trials for metastatic and advanced primary tumors, respectively, in breast cancer55. Moreover, blocking antibodies with GSI activity have also been developed. These agents target the components of GSI complex, such as nicastrin, the largest member of the complex. Antinicastrin monoclonal antibodies clone 2H6 elicited pleiotropic antimetastatic activities on invasive cancer cell lines, including an attenuation of invadopodia degradation of the extracellular matrix and delayed cancer cell extravasation through endothelial cells in thein vitro Boyden chamber invasion assay57.
The enthusiasm for anticancer and antimetastatic application of GSIs is mitigated by the limitation to gamma secretase targeting, considering that this Notch signaling component is not always critical in the pathological phenotype of malignancies. In addition, a number of tissues physiologically require Notch activity for tissue plasticity. Gastric epithelium is the most relevant example of such tissue. Its functional alterations partly explain the gastrointestinal toxicity observed following GSI treatment53,55,56. In vivo experiments suggest that currently, antinicastrin monoclonal antibodies are more potent than GSIs in clinical trials, with minimally marked gastrointestinal signs57. Collectively, these observations suggested the possibility to improve the therapeutic outcome of the targeting of Notch signaling by changing the specific targets and the approaches used. These findings raised the question of whether developing the inhibitors for specific Notch receptors or ligands would also improve therapeutic outcome.
Targeting Notch ligands and receptors
Notch ligand and receptor targeting is a particularly interesting approach because it allows specific targeting of Notch receptors or ligands critical in the pathological phenotype of malignancies, including the metastatic phenotype. For example, Notch1 was reported to control metastatic processes in small cell lung cancer cell lines and to initiate EMT and invasion of breast cancer cells48,58. The targeting of blocking monoclonal antibodies, specifically Notch receptor subtypes, has been developed. These molecules mainly act on the EGF-like repeats of Notch receptors. Various Notch receptor blocking antibodies have entered clinical trials for metastatic and advanced solid cancers. For example, tarextumab (OMP-59R5), an anti-Notch2/3 receptor currently in phase 1 trials in patients with untreated metastatic pancreatic cancer, is showing promising therapeutic effects with the antineoplastic (chemotherapy) drugs Nab-Paclitaxel and Gemcitabine59. Examples of other Notch receptor or ligand blocking antibodies currently in clinical trials include Notch1 monoclonal antibody OMP-52M5 and the anti-DLL4 demcizumab (OMP-21M18)55.
Besides, small molecules acting as receptor or ligand decoys were also developed. A recent report by Kangsamaksin and collaborators10 provided experimental evidence for the potential of decoy molecules for silencing the Notch receptors and ligands specifically60. These authors developed Notch decoys N1-13 and N1-24, interfering with Dll-Notch and Jagged-Notch interactions, respectively. The decoy molecules potently disrupted tumor growth and promoted normal endothelial sprouting in tumor microenvironment by reducing angiogenic sprouting, vessel perfusion and pericyte coverage, and other pro-angiogenic processes54. Besides the possibility to improve the therapeutic outcome, Notch receptor or ligand-specific targeting may also decrease the severity or abrogate adverse gastrointestinal effects observed with the unspecific targeting of Notch signaling.
Antimetastatic potential of Wnt and Hedgehog targeting
Wnt targeting
Wnt signaling and cancer
The Wnt family of signaling molecules encompasses a number of cysteine-rich glycosylated secreted ligands that bind to the extracellular domain of frizzled family of receptors. Wnt binding triggers a signaling cascade resulting in the activation of genes involved in stem cell maintenance, cell survival, proliferation, motility, migration, and fate determination during the development. Wnt aberrant overexpression can activate the pathogenic developmental-like Wnt signaling activity in transforming cells, favoring stemness and chemotherapy resistance61–63.
The canonical Wnt/β-catenin pathway is the most studied Wnt signaling pathway. In the canonical pathway, Wnt binding to frizzled-7 leads to the disaggregation of β-catenin destruction complex made of the tumor suppressor adenomatous polyposis coli (APC), the serine/threonine protein kinase glycogen synthase kinase 3, and casein kinase. Canonical Wnt/β-catenin pathway requires the co-receptors LRP5 and LRP6. The disaggregation of β-catenin destruction complex results in the inhibition of β-catenin destruction, and conversely, in the cytoplasmic accumulation of β-catenin. When in sufficiently high levels, a fraction of cytoplasmic β-catenin migrates to the nucleus where it interacts with a member of the TCF/LEF-1 family of transcription factors. Consequently, these transcription factors activate gene encoding for proteins involved in EMT, survival, angiogenesis motility, and invasion, such as c-Myc, Jagged1, VEGF, CCL2, snail, slug, vimentin, and metalloproteinases62–67.
Two other Wnt signaling pathways, i.e., the non-canonical Wnt pathways, namely, the planar cell polarity and Wnt/calcium pathway, were also described. The Wnt non-canonical pathway does not require the co-receptors LRP5/6. The non-canonical planar polarity pathway activates small Rho GTPases, such as RAC1, Rho, and Cdc42, which control actin dynamic through Rho kinase, the actin-binding protein cofilin, and the MAPK kinase JNK68–71.
The Wnt/calcium pathway, which is initiated by non-canonical Wnt ligands, such as Wnt5a, activates phosphoinositide phospholipase C (PLC) and phosphodiesterase 6 (PDE6). The PLC-mediated cleavage of the plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate releases inositol trisphosphate and diacylglycerol. The resulting signaling cascades induce the release of mitochondrial Ca2+ in the cytoplasm, with subsequent activation of Ca2+-dependent enzymes, such as Ca2+/calmodulin-dependent kinase II, protein kinase C (PKC), and the serine/threonine protein phosphatase calcineurin68–71.
Role of Wnt signaling in metastasis and therapeutic potential
Evidence sustaining a role for aberrant Wnt pathway activation in metastasis was provided by reports of the capability of Wnt antagonism to suppress tumorigenesis and metastasis partly (i) by enhancing the expression of epithelial marker, such as E-cadherin and keratin 8/18 (preventing EMT) and (ii) by decreasing the expression of pro-metastatic factors, such as slug, twist, snail, and metalloproteinases8–10. The aberrant accumulation of β-catenin in the cytoplasm observed in colorectal tumors and many other solid cancers results from tumor-promoting epigenetic events, such as DNA demethylation on aCTNNB1 gene promoter that activates β-catenin synthesis, and DNA hypermethylation on an APC gene promoter that silence this gene, resulting in the decrease of cellular levels of β-catenin destruction complex member APC70,71. Similarly, the absence of degrading the β-catenin observed in many cancers may also result from the silencing mutations of APC gene, whereas the activating mutations of CTNNB1 gene may also increase the β-catenin cellular levels72,73. The genetic and epigenetic suppression of the other members of the destruction complex was also reported74,75. Furthermore, a fraction of β-catenin is bound to the E-cadherin in the cytoskeleton, thus the EMT-associated loss of E-cadherin results in the release of β-catenin76. The failure of misshaped β-catenin (product of mutations) to bind to E-cadherin also increases the β-catenin cytoplasmic levels.
The canonical Wnt3 signaling consistently promoted a partial EMT-like transition with increased N-cadherin, twist, slug, and decreased E-cadherin in the trastuzumab-resistant breast tumor cells, and the Wnt3 knockdown by siRNA decreased the expression of EMT triggering factor EGFR in these malignant cells77. In addition, activating the Wnt /β-catenin signaling correlates with EMT and the potency of proliferation and invasiveness in prostate cancer cells63,78. The nuclear localization of β-catenin is a molecular marker of EMT in colon cancer 79. The inhibition of Wnt signaling with β-catenin shRNA reversed HIF-1a-induced EMT in human prostate cancer63,78. Moreover, Wnt/β-catenin may promote EMT via direct transactivation of EMT transcription factors, such as ZEB131.
The non-canonical Wnt signaling pathways also played pivotal roles in cancer metastasis. For example, the well-known activator of non-canonical Wnt pathway Wnt5a increased not only the metastasis in a PKC- and a Stat3-dependent manner in melanoma but also the expression of melanoma immunogens and severity markers GP100, MART-1, and tyrosinase80,81.
Wnt5a is upregulated in epithelial tumors, where it promotes EMT and migration in a β-catenin-independent manner82–89. The treatment of these tumor cells with Wnt5a resulted in an upregulation of EMT stimulating factors, including vimentin, Snail1, and slug82. In addition, Gujral and collaborators104 reported an upregulation of Wnt5 receptor Frizzled2 in metastatic liver, lung, colon, and breast cancer cell lines and in high-grade tumors. Such expression correlated with the overexpression of EMT markers. In the same study, pharmacological and genetic perturbation analyses suggested that Wnt5a/Frizzled2 signaling induces EMT and cell migration via a previously unrecognized Fyn/Stat3-dependent non-canonical pathway.
Besides, considering the involvement of planar polarity pathway in regulating actin cytoskeleton mediated via its capability to activate the Rho GTPases (Section 2.3.1)68–70, a role for this non-canonical Wnt pathway in the cytoskeleton reorganization supporting motility, migration, and invasion can be predicted. The experimental evidence suggests the involvement of this pathway in cellular motility and the invasive capacity of metastasis-promoting cells in various cancers, including pancreatic ductal adenocarcinoma and breast cancer85,86. Moreover, Frizzled receptors that can trigger canonical and non-canonical signaling, such as Frizzled2 and Frizzled7, reported the capability to interact with Rho-A and other small GTPases during metastatic cascade. For example, Wnt3a/Frizzled2 signaling stimulates cell migration and invasion in a Rho-A-dependent manner in multiple myeloma83,84, and Wnt5a was reported to promote cell migration in breast cancer through the Dishevelled2/Daam1/Rho-A axis89.
The canonical Wnt signaling pathway inhibitors, such as the CBP/β-catenin antagonist PRI-724, have been developed. The pivotal roles of this signaling pathway in maintaining the stemness of tissue stem cells and in regulating the tissue homeostasis and cell fate along the lifespan raise concerns for safety of Wnt-based anticancer strategies despite the potential of Wnt signaling targeting for cancer stem cell elimination90,91 and metastasis suppression, discouraging their development. Preclinical studies91 and phase 1 clinical trials92 suggested that PRI-724 is relatively well-tolerated, and the drug is currently in phase 2 trials combined with chemotherapy drugs and the angiogenesis inhibitor bevacizumab (NIH trial IDs 3C-13-3, NCI-2015-00436, and NCT02413853). Experimental evidence suggests differences in Wnt signaling mediating stemness in malignant and non-malignant cells93. The characterization of such differences may open new avenues for safe targeting of Wnt signaling.
Hedgehog, metastatic cascade, and emerging therapies
Hedgehog family
The vertebrate Hedgehog family of lipid-modified proteins encompasses Desert Hedgehog, Indian Hedgehog, and Shh. Shh is the most studied Hedgehog family member. Hedgehog proteins are the ligand of 12-pass transmembrane protein receptors, namely, patched. In the absence of interactions with ligands, patched receptors catalytically maintain the 7-pass transmembrane protein smoothened (Smo) in an inactive state. The inhibitory activity of patched is abrogated upon ligand binding, and Smo activates the transcription factors of Gli zinc-finger protein family in the cytoplasm. Then, Gli proteins migrate to the nucleus where they transactivate or repress various genes94,95.
Hedgehog played pivotal roles in cancer development96 and in metastatic cancer. For example, the loss and gain and pharmacological inhibitions of function experiments suggested that Hedgehog signaling is required along the metastatic cascade97–101. Examples include studies showing (i) that high Hedgehog-Gli signatures coincide with the development of metastases in colon cancer98 and (ii) that Gli1 transcription factor activity links with tumor aggressiveness in papillary thyroid cancer97. In addition, Hedgehog promotes EMT in various solid tumors, including pancreatic, colon, and breast cancers99. Gli proteins are involved in the TGF-β mediated EMT in hepatocellular carcinoma, marked by an increased expression of Snail1101. The Gli1 knockdown resulted in decreased hepatocellular carcinoma migration and invasion101. Shh enhanced the gastric cancer cell motility and invasiveness, whereas no increase was observed in cells treated with Shh signaling inhibitor cyclopamine-KAAD or anti-Shh monoclonal antibodies100. Moreover, Yoo and collaborators105 demonstrated that Shh signaling promotes gastric cancer metastasis at least partly through the activation of PI3K/Akt signaling-mediated matrix metallopeptidase 9 and EMT.
Hedgehog-based therapeutic strategies
The Smo inhibitors are the largest group of Hedgehog signaling inhibitors. The naturally occurring alkaloid cyclopamine is a typical example. Cyclopamine inhibits the growth and invasiveness of a tumor cell in vitro and in vivo103–105. However, the translation to cancer treatment has been challenging due to the poor stability and solubility of cyclopamine and also due to its severe adverse effects resulting from the activity of the products of its metabolism101,106. Soluble and stable derivatives of cyclopamine were developed, including (i) the emerging prodrug cyclopamine glucuronide tested in glioblastoma multiform models107 and (ii) vismodegib (GDC-0449), currently in phase 2 clinical trials in patients with metastatic pancreatic adenocarcinoma108, advanced primary tumors and metastatic basal cell carcinoma109, and pediatric brain tumors110. Other Smo inhibitors, currently in clinical trials for refractory and metastatic cancer treatment, include IPI-926111, LDE225112, BMS-833923113, and PF-0449913114. Most of these prodrugs and drugs have an acceptable safety profile with only low grade or relatively minor adverse effects with promising anticancer and antimetastatic effects. Moreover, Gli inhibitors, another class of Hedgehog inhibitors, include prodrugs, such as Gant-58 and Gant-61, which displayed a strong antimetastatic potential in preclinical studies100–123.
A number of blocking antibodies targeting Hedgehog signaling are available, while only a few inhibitors of Hedgehog ligands were developed. The monoclonal antibody 5E1, one of the most studied Hedgehog-targeting antibodies, antagonizes the three Hedgehog ligands. Studies in nude mice revealed 5E1 capable of inhibiting the metastasis of pancreatic cancer118. The Hedgehog ligand inhibitor robotnikinin was discovered by Stanton and collaborators102 using approaches for small molecule microarrays and diversity-oriented synthesis. This small molecule mediates extracellular Shh inhibition by binding to this protein, preventing interactions with patched receptors. Data on robotnikinin effects on metastatic processes are lacking.
Conclusions
Targeted therapies based on emerging master developmental pathways represent promising approaches for metastasis prevention and cancer stem cell elimination. Such therapeutic potential is due to the tumorigenesis-mediated hijacking of EMT, cell motility, migration, stemness, angiogenesis, and other key phenomena determining cell fate controlled by developmental pathways. These phenomena are pivotal for all steps of solid tumor initiation, maintenance, and development. Various classes of Notch signaling inhibitors are currently in clinical trials. The development of therapeutics targeting specifically the Notch signaling components critical for malignancy pathological phenotype is expected to improve the therapeutic outcome and decrease adverse effects. Wnt signaling targeting, in which safety is a serious concern, is surprisingly well-tolerated in clinical trials and is expected to be a game changer for cancer stem cell elimination and metastasis prevention. Unlike Notch and Wnt pathways, translating the Shh signaling blockade to cancer treatment has been challenging because of severe adverse effects and poor therapeutic potential of various monoclonal antibodies and naturally occurring Smo inhibitors. Further mechanistic studies of the involvement of developmental pathways in refractory and metastatic cancers may further improve the targeting efficiency and the therapeutic outcome.
Footnotes
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received March 22, 2016.
- Accepted March 27, 2017.
- Copyright: © 2017, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.
- 20.↵
- 21.↵
- 22.↵
- 23.
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.
- 65.
- 66.
- 67.↵
- 68.↵
- 69.
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.
- 88.
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.
- 116.
- 117.
- 118.↵
- 119.
- 120.
- 121.
- 122.
- 123.↵
- 124.
- 125.
- 126.
In this issue






{kind=link}
Jump to section
Related Articles
Cited By...
- Targeted mechanical forces enhance the effects of tumor immunotherapy by regulating immune cells in the tumor microenvironment
- Role of the mechanical microenvironment in cancer development and progression
- Sex-determining region Y box-containing genes: regulators and biomarkers in gynecological cancers
- Cancer stem-like cells directly participate in vasculogenic mimicry channels in triple-negative breast cancer