

Abstract
Liver cancer, primarily hepatocellular carcinoma (HCC), is a major cause of cancer-related death worldwide. HCC is a suitable model of inflammation-induced cancer because more than 90% of HCC cases are caused by liver damage and chronic inflammation. Several inflammatory response pathways, such as NF-κB and JAK/STAT3 signaling pathways, play roles in the crosstalk between inflammation and HCC. MicroRNAs (miRNAs) are evolutionarily conserved, short endogenous, non-coding single-stranded RNAs that are involved in various biological and pathological processes by regulating gene expression and protein translation. Evidence showed that miRNAs play a pivotal role in hepatitis virus infection and serve as promoters or inhibitors of inflammatory response. Aberrant miRNA was observed during liver inflammation and HCC. Many dysregulated miRNAs modulate the initiation and progression of inflammation-induced HCC. This review summarizes the role and functions of miRNAs in inflammation-associated HCC, as well as the designed therapeutics targeting miRNAs to treat liver inflammation and HCC.
keywords
Inflammation and liver cancer
The implication of inflammation in the development of cancers is well recognized. In the 19th century, Virchow observed leukocytes in tissues that contained inflammatory infiltrates1, which later on was confirmed in several clinical cases2. However, these insights were not further examined until data collected from epidemiological studies and experimental models showed a link between chronic inflammation and various types of cancers3,4. Chronic inflammation occurs with continuous inflammatory signaling caused by viral, bacterial, and parasitic infections or chemical irritants; exposure to chemical irritants results in recruitment of inflammatory cells, increased production of reactive oxygen species (ROS), and reduced DNA repair5,6. Most cancer tissues show specific types of inflammatory response; this subset accounts for approximately 20% of human cancers worldwide and is considered an essential component of tumorigenesis, including tumor initiation, progression, growth, and metastasis5,7.
Liver cancer is one of the most difficult cancers to treat and is the second leading cause of cancer death worldwide, especially in China, where it accounts for half of total cancer incidence and death8. Liver cancer is strongly correlated with viral hepatitis infection; more than 90% of liver cancers are associated with chronic hepatitis or liver cirrhosis9. Inflammation plays an essential role in responding to harmful stimuli and insults. However, cell death-regeneration cycles are driven by persistent inflammation that occurs when inflammation-associated signaling cannot be inhibited and subsequently results in chronic injury.
Many factors provoke an inflammatory response in the liver and are well known risk factors for liver cancer. Liver cancer occurs during the progression of many chronic liver diseases associated with inflammation, such as hepatitis and steatohepatitis10. Hepatitis B virus (HBV) and hepatitis C virus (HCV) are found in approximately 80%-90% of hepatocellular carcinoma (HCC) patients11. HBV is an enveloped DNA virus with double-stranded circular DNA. HBV integrates into the host genome, which results in genomic instability12. Chronic HBV infection is the dominant risk factor for liver cancers in Asia and Sub-Saharan Africa, particularly in China. HCV is a single-stranded RNA virus that interacts with numerous proteins in hepatocytes and promotes the progression of hepatocarcinogenesis13. Chronic HCV infection is the dominant risk factor for liver cancers in Japan, United States, and Europe. Cirrhosis contributes to hepatocarcinogenesis and is observed in most HCC patients with HBV or HCV infection14.
In addition to these viral infections, acetaldehyde and other metabolites generated by alcohol metabolism increase the amount of ROS and reduce DNA repair, which leads to HCC15. Epidemiological analysis revealed that alcohol consumption is positively associated with HCC incidence; diabetes is another risk factor for HCC because epidemiologic studies reported that patients with type 2 diabetes have nearly 2.5-fold increased risk for HCC16,17. Recently, obesity was defined as a risk factor with an important role in HCC development18. Inflammation induced by viral or non-viral infection promotes the initiation and progression of HCC; HCC develops in the background of chronic hepatitis19.
Several signaling pathways link inflammation and cancer. Constitutive activation of these inflammatory signaling pathways, such as NF-κB and JAK/STAT signaling pathways, occurs during the process of inflammation and is observed in various cancers. The first link between NF-κB and cancer was revealed in a study showing that cancer cells were more sensitive to apoptosis-inducing stimuli after the NF-κB signaling pathway was blocked20. Over the past decades, the diverse roles of NF-κB signaling have been discovered in various types of carcinomas, including head and neck squamous cell carcinoma, leukemia, and cancers of breast, liver, prostate, colon, and lung21. Constitutive NF-κB activation was observed in various cancers, where NF-κB induces the expression of many pro-survival and anti-apoptotic factors, such as c-IAP, XIAP, and members of the Bcl-2 family of proteins22,23. Analogously, the activation of NF-κB might downregulate the expression of tumor suppressors, such as phosphatase and tensin homolog (PTEN); the downregulated expression increases Akt signaling and suppresses the apoptosis of cancer cells24. In addition to its pro-survival function, NF-κB promotes cell cycle progression by inducing cyclin D1 and c-myc expression25,26. NF-κB signaling can also induce molecules such as vascular endothelial growth factor (VEGF) and matrix metalloproteinases, and NF-κB can promote angiogenesis and invasion by driving the expression of these proteins in many cancers27. In 2004, a group from Israel confirmed that NF-κB is an effective therapeutic target to treat HCC28. HCC development in a mouse model was prevented through NF-κB inhibition by a modified form of IκB called super-repressor IκB.
Another well-known signaling pathway that links inflammation and cancer is the Janus kinase (JAK)/signal transducer and activator of transcription (STAT). This signaling pathway is also constitutively activated in most cancers. STAT3, one of the most famous STAT family members, was identified as a transcription factor that binds to the IL-6-responsive element in hepatocytes29. This binding suggests that STAT3 is a key regulator of inflammation. However, inflammatory cytokines are not the only factors that activate STAT3 signaling. Numerous studies reported that STAT3 can also be activated by oncogenic proteins including Ras or Src30,31. Carcinogens, such as polychlorinated biphenyls, promotes liver cancer and regulates the activity of STAT3 in HCC and normal liver cells32. Therefore, STAT3 is a key role in inflammation regulation and cancer development. Likewise, the constitutive activation of STAT3 leads to the expression of Bcl-2 family proteins essential for survival, such as survivin and cIAP2; moreover, the apoptosis of cancer cells occurs when STAT3 is downregulated in tumors33–36. STAT3 promotes the proliferation of cancer cells by inducing cyclin D1 and c-myc expression37,38. In addition to molecules associated with proliferation and apoptotic inhibition, STAT3 regulates the expression of the metalloproteinase family and VEGF, consequently promoting angiogenesis and metastasis39. STAT3 is also activated in hepatocytes following interleukin stimulation. STAT3 is a key regulator of inflammation and cancer and thus might be an effective therapeutic target in inflammation-induced cancer. In 2006, Li et al.40 inhibited STAT3 expression with antisense oligonucleotides in HCC cells and found that STAT3 greatly reduced cell proliferation and migratory potential as well as induced apoptosis.
MiRNAs are a novel class of molecules linking inflammation and HCC
MiRNAs constitute a class of short endogenous non-coding RNAs that are highly conserved throughout evolution. The biogenesis of miRNAs requires multiple steps. The majority of miRNAs are transcribed in the nucleus by RNA polymerase II as a primary miRNA (pri-miRNA); the typical pri-miRNA contains stem-loop structures. Furthermore, these stem-loop structures are cleaved into precursor miRNAs (pre-miRNAs) by Drosha, an RNA-specific ribonuclease enzyme complex. Dicer functions in the biogenesis of miRNAs as pre-miRNAs are transported from the nucleus into the cytoplasm by Exportin 541. Dicer is an endonuclease that specifically recognizes double-stranded RNA complexes and transforms the pre-miRNA into a 20-22 nt miRNA duplex42. The mature miRNA is incorporated into an RNA-induced silencing complex (RISC) through its interaction with Ago proteins; this miRNA recognizes and binds to the 3’-UTR of target mRNAs43.
The first miRNA was discovered in Caenorhabditis elegans in 1993 and was identified as an essential regulator of postembryonic developmental events44. From that first discovery, additional miRNAs have been identified and investigated in multiple organisms. Currently, miRNAs are recognized as key mediators influencing nearly all biological processes, including development, immune response, differentiation, proliferation, apoptosis, invasion, and metastasis. In addition, the expression of numerous miRNAs is dysregulated during nearly all pathological processes. MiRNAs gained a widely accepted role in initiation and progression of various human cancers, including HCC; evidence indicated that these miRNAs act as oncogenes or tumor suppressors45,46. Multiple miRNAs are upregulated or downregulated during hepatocarcinogenesis. Some of these miRNAs might be significant to diagnosis or prognosis of HCC patients47. Numerous miRNAs are reported to be aberrantly expressed during hepatitis and hepatocellular carcinoma. Accumulating evidence revealed that miRNAs are a novel class of molecules linking inflammation and HCC.
MiRNAs associated with hepatitis virus infection and HCC
Chronic hepatitis virus infection leads to liver inflammation, damage, fibrosis, cell regeneration, and malignant transformation (HCC). Many miRNAs play a crucial role in the regulation of hepatitis virus-induced chronic infla-mmation and HCC (Table 1). For instance, miR-27 is upregulated by HCV infection and can promote cell migration and invasion by suppressing Spry2 in HCC cells106. MiR-146a expression is consistently increased in HCV-infected cells, primary hepatocytes, and liver tissue from HCV-infected patients. Increased miR-146a level promotes viral infection and is associated with the pathogenesis of liver disease107. In the following section, we will summarize the key roles of relevant miRNAs in hepatitis virus infection and HCC.
MiRNAs associated with hepatitis virus infection and HCC
Let-7
The first miRNA discovered in humans is let-7, which is highly conserved across animal species and promotes differentiation108,109. Recent studies revealed that let-7 is frequently downregulated in many cancers, such as breast, ovarian, prostate, lung, and liver cancers110. The HBV X protein (HBx) encoded by the HBV genome is a multifunctional transactivator that plays crucial roles in HBV-associated hepatocarcinogenesis. HBx lacks a DNA binding domain and acts as a trans-coactivator through interaction with other cellular transcription factors, by which HBx might regulate let-7 family members. In addition to HBx induction, HBV preS2 RNA can sequester let-7g, which in turn reduces intrinsic let-7g functions in HCC111. Wang et al.48 concluded that HBx-mediated down-regulation of let-7 might be responsible for reduced let-7 expression in HBV-associated HCC. MiRNAs from the let-7 family act as tumor suppressors in HCC by inhibiting proliferation and invasion, as well as inducing apoptosis by targeting many oncogenes including high mobility group proteins, Bcl-xL, STAT3, and c-myc51,112,113. Further clinical research suggested that let-7 expression level is associated with poor differentiation114.
MiR-122
MiR-122 is the most abundant miRNA expressed in the liver, which accounts for 70% of the total miRNAs in an adult liver115. The expression of miR-122 is increased in the liver during embryonic development, and the first identified target of this miRNA is cationic amino acid transporter 1116. In recent years, increasing evidence suggested that miR-122 plays an important role in initiation and progression of HCC. Li et al.117 found that miR-122 expression is low in patients with chronic hepatitis B and HCC tumors and has negative correlation with fibrotic stage, age, and liver transaminase levels. The downregulation of miR-122 in patients with chronic hepatitis B and HCC tissues is affected by HBV infection. HBx overexpression reduces the expression of miR-122118. PPARγ was identified as a transcription factor regulating miR-122 expression; the underlying cause of HBx-induced miR-122 downregulation might be the direct interaction between HBx and PPARγ, which blocks PPARγ nuclear translocation119. In 2013, Li et al.120 further reported that all four HBV mRNAs contain a binding site for miR-122 and act as a miRNA sponge sequestering miR-122 in HBV-infected cells. In addition, miR-122 knockout mice will possibly develop hepatitis, fibrosis, and HCC. The reduced miR-122 expression in HCC tissues is characterized by a poor prognosis and consistent with a suppressed hepatic phenotype and an increased metastatic potential121. Moreover, miR-122 expression can be suppressed by two inflammatory cytokines, IL-6 and TNF-α, which downregulate the miR-122 transcription factors C/EBPα and HNF3β and induce c-myc-mediated C/EBPα inhibition in chronic hepatic inflammation117. MiR-122 downregulation results in the upregulation of target genes that promote hepatocarcinogenesis, including pituitary tumor trans-forming gene binding factor (PBF), cyclin G1, PKM2, ADAM10, ADAM17, SRF, and PKR activator122–124.
MiR-101
MiR-101 is another miRNA that is downregulated by HBV infection125. HBV downregulates miR-101 expression by inhibiting its promoter activity. MiR-101 downregulation was reported in various cancers, including HCC, suggesting that miR-101 functions as a tumor suppressor52,126. Reduced miR-101 expression in HCC tissues is associated with advanced tumor progression and poor survival in HCC patients; the serum miR-101 level might be a potential HCC biomarker127,128. Overexpression of miR-101 sensitizes the HCC cells to apoptosis induced by serum deprivation and chemotherapeutic drugs. Mcl-1, a member of the Bcl-2 family, is a target of miR-101 and sensitizes cancer cells to apoptosis52. MiR-101 enhances cisplatin-induced apoptosis by targeting STMN1, RAB5A, and mTOR, as well as suppresses epithelial-mesenchymal transition (EMT) in hepatocytes via the regulation of ZEB1 expression, which results in E-cadherin down-regulation129,130.
MiR-155
In 2010, the first link between miR-155 and hepatitis viruses was based on the observation that miR-155 precursor (termed BIC) is responsible for HCV infection131. After patients with hepatitis C were treated with interferon alpha and ribavirin, the lowest BIC expression was detected in patients who lacked HCV RNA in both serum and peripheral blood mononuclear cells. Previously, miR-155 was identified as an onco-miR in various cancers, including lung, breast, stomach, and prostate cancers132,133. A mouse model revealed that miR-155 induced by inflammation could promote autoimmune inflammation by enhancing inflammatory T cell development134. MiR-155 up-regulation occurs before HCC carcinogenesis. A positive correlation was observed between miR-155 expression and IP-10 levels in the plasma, which is widely considered a marker of liver inflammation degree104. Upon NF-κB activation by HCV-RNA infection, miR-155 expression is induced. The upregulation of miR-155 in cases of hepatitis and HCC promotes hepatocyte proliferation and HCC tumor growth by activating the Wnt/β-catenin signaling pathway. In addition to HCC pathogenesis during chronic viral infections such as HCV, the upregulation of miR-155 was also observed in the pathogenesis of non-alcoholic steatohepatitis (NASH)-associated HCC. However, similar to HCV-induced miR-155, the activation of NF-κB signaling pathway plays a key role in miR-155 expression in HCC and other hepatocyte-derived cells. In a mouse model of NASH, miR-155 downregulated the tumor suppressor target C/EBPβ, thus promoting liver pathogenesis132.
MiR-21
MiR-21 is a well-established onco-miR that is upregulated in various cancers, including lung, breast, prostate, colon, stomach, pancreatic, and liver cancers135,136. The relationship between miR-21 and inflammation was observed in UV-exposed skin, where miR-21 was activated by PPAR via the TGF-β signaling pathway137. The upregulation of miR-21 was reported in other inflammation-associated tissues and cancers, such as allergy-induced airway inflammation, gastric cancer, pre-B-cell lymphoma, and colorectal cancer138–141. The relevance of miR-21 and inflammation in liver tissues was established by associating biomarkers with HCC diagnosis and prognosis. MiR-21 is expressed in tumor cells and tumor-associated fibroblasts. In 2010, miR-21 and miR-122 were analyzed in tissues obtained by needle biopsies from both HCV-infected and non-infected patients. The expression of miR-21 was positively correlated with fibrotic stage in patients with chronic hepatitis C; miR-21 promotes liver fibrosis by targeting the fibrosis inhibitor SMAD7135. In serum, miR-21 was significantly upregulated in patients with either chronic hepatitis B or HCC142. In addition, one study reported that HBx increases the expression of miR-21 and subsequently promotes the progression of HCC by targeting PTEN143. In another study, HBx-mediated miR-21 upregulation inhibits expression of the tumor suppressor PDCD4 in HCC cells144.
MiR-221
MiR-221 is another onco-miR that is upregulated in cancers such as breast, prostate, glioma, and liver cancers46,145–147. Moreover, the expression of miR-221 in later stages of HCC is higher than that in early stages148. Therefore, miR-221 is a poor prognosis risk factor because it was increased in mouse models of liver fibrosis and upregulated in the human liver in a fibrosis progression-dependent manner149,150. Overex-pression of miR-221 in isolated and purified primary hepatocytes increased cell proliferation151. HBx promotes the proliferation of HCC cells and upregulates the expression of miR-221 during both HBV infection and HCC98. HCV infection can also induce the upregulation of miR-22199. Several tumor-related genes were reported to be the targets of miR-221; among these genes, p27Kip1 is well-recognized147. The expression of miR-221/p27Kip1 axis was confirmed in glioblastomas, breast, lung, and liver cancers152–155. Other than p27Kip1, many miR-221 targets were reported, such as HDAC6, PTEN, and tissue inhibitor of metalloproteinase-3 (TIMP3)156,157. In mouse models of HCC, tumor-bearing mice that were treated with chol-anti-miR-221 have a survival advantage compared with mice in the control group. MiR-221 is a potential target for HCC treatment and functions by inhibiting the progression of HCC and promoting survival158. MiR-221 transgenic animal models exhibited a strong predisposition toward the development of HCC159.
MiRNAs as regulators of inflammatory signaling in HCC
Chronic inflammation can facilitate the development and progression of HCC by the constitutive activation of inflammation signaling pathways. Aberrant activation of these pathways promotes inflammation-associated hepatocarcinogenesis. Inhibition of these signaling pathways would be a novel therapeutic strategy to suppress this inflammation-associated process. Many miRNAs were reported to regulate the inflammatory signaling mediated by NF-κB and JAK/STAT3, among others, in HCC (Table 2). Our group screened several miRNAs that might suppress NF-κB activation. Among these miRNAs, miR-195, miR-194, and miR-127 suppressed NF-κB activation by directly or indirectly targeting NF-κB signaling; these miRNAs also function as tumor suppressors in HCC160,162,163. MiR-219 is reduced in HCC where it functions as a tumor suppressor; this miRNA might suppress JAK/STAT3 signaling via direct targeting of structural maintenance of chromosome 4 protein (SMC4)183. MiR-637 is a tumor suppressor that inhibits cell growth and induces apoptosis by suppressing STAT3 activation in HCC186. Some typical miRNAs that behave as regulators in inflammatory signaling are summarized below.
MiRNAs as regulators of inflammatory signaling in HCC
MiR-26a
MiR-26a was suggested to play an important role in cirrhosis and inflammation-induced HCC188. Reduced miR-26a expression was detected in HCC tissues; the decreased expression in HCC tissues might be an independent predictor of poor survival189. Clinical analysis revealed that the expression of miR-26a is negatively correlated with IL-6 mRNA and IL-6 protein level. As IL-6 is a direct target of miR-26a, the mRNA and protein levels of this cytokine are significantly reduced by miR-26a overexpression in HCC cells. MiR-26a functions as a tumor suppressor that inhibits tumor growth and metastasis, partly through the IL-STAT3 signaling pathway184. Furthermore, miR-26a regulates the recruitment of macrophages190. M-CSF expression was also reported as a predictor of frequent metastasis and poor survival; miR-26a affects macrophage polarization and inhibits macrophage recruitment by down-regulating M-CSF expression191. In addition, HCC cells transfected with miR-126b exhibit significantly reduced NF-κB reporter activity in response to TNFα. Two well-known upstream regulators of NF-κB signaling pathway, TAK1 and TAB 3, were identified as direct targets of miR-26b. MiR-26b prevents doxorubicin-induced NF-κB activation and promotes apoptosis in HCC cells164.
MiR-451
In one study, miR-451 was often downregulated in primary HCC tissues; reduced miR-451 expression was significantly associated with advanced TNM stage, lymph node metastasis, vascular invasion, and poor survival, indicating that miR-451 might act as a tumor suppressor in HCC192. MiR-451 might inhibit HCC tumorigenesis and metastasis by down-regulating cyclin D1 and c-myc at both mRNA and protein levels. Further study indicated that miR-451 overexpression affected the NF-κB nuclear translocation and significantly decreased NF-κB activity by directly targeting IKKβ. As expected, the inhibition of miR-451 significantly increased NF-κB activity in HCC cells176. Moreover, a recent study revealed that reduced expression of the IL-6 receptor was observed in cells that overexpressed miR-451, and that IL-6 receptor was a direct target of miR-451. MiR-451 reduced VEGF production in HCC cells by targeting the IL-6/STAT3 signaling pathway177.
MiR-124
MiR-124 is a well-known tumor suppressor that is frequently downregulated in various cancers. Numerous studies revealed that miR-124 inhibits inflammatory signaling by targeting STAT3 in different cancer types including glioblastoma, gastric cancer, endometrial cancer, colon cancer, and HCC179,193–196. Recently, NF-κB p65 was reported to be a direct miR-124 target in B-cell lymphomas. The downregulation of miR-124 in B-cell lymphomas enhances the NF-κB signaling pathway and upregulates genes, such as myc and Bcl-2, associated with proliferation, anti-apoptosis, metastasis, and pro-survival197. STAT3 was identified as a direct miR-124 target in HCC; one study found that miR-124 inhibited cell proliferation and induced apoptosis in HCC cells by targeting STAT3179. Moreover, an HNF4α-NF-κB feedback circuit that includes miR-124 was identified in HCC. Reduced HNF4α could be a biomarker for poor prognosis, as HNF4α suppresses HCC metastasis by inhibiting NF-κB activation. MiR-124 also plays an important role in RelA suppression by HNF4α. MiR-124 was identified as an HNF4α target that binds to the 3’UTR of RelA. MiR-124 significantly downregulates RelA expression, represses NF-κB activation, and inhibits HCC tumorigenesis198.
MiR-125b
MiR-125b is a well-recognized miRNA that participates in innate immune response199. Our group was the first to reveal the role of miR-125b in HCC. We discovered that miR-125b was significantly downregulated in HCC, could be used as a biomarker for prolonged HCC patient survival, and functions as a tumor suppressor47. MiR-125b inhibits the proliferation of HCC cells by inducing cell cycle arrest during G1/S transition via its target gene, Lin28B200. Zhao et al.201 found that miR-125b induced HCC apoptosis by suppressing Bcl-2. IL-6R was identified as a direct target of miR-125b. IL-6R was characterized as the subunit that binds to IL-6; the binding of IL-6 to IL-6R activates the JAK/STAT3 signaling pathway. MiR-125b can block the IL-6-induced phosphory-lation and nuclear translocation of STAT3 in HCC cells, which in turn promotes HCC apoptosis180,202. In addition, miR-125b serves as a tumor suppressor that inhibits migration and invasion of HCC cells by targeting the transcriptional co-activator with PDZ-binding motif (TAZ). MiR-125b can also suppress EMT by downregulating SMAD2 and SMAD4203,204. EZH2 was also associated with miR-125b downregulation205.
MiR-370
The role of miR-370 in carcinogenesis is controversial. Several studies revealed that miR-370 acts as tumor promoter in gastric cancer, prostate cancer, and AML; whereas other studies reported miR-370 as a tumor suppressor in leukemia and oral squamous carcinoma206–210. Xu et al.167 observed that the down-regulation of miR-370 was associated with HCC development in rats and correlated with poor survival in HCC patients, indicating that miR-370 serves as a tumor suppressor in HCC. The restoration of miR-370 inhibits proliferation, migration, and invasion of HCC cells and suppresses tumorigenesis of HCC by directly targeting Lin28A. Lin28A is a post-transcriptional modulator of mRNAs that can directly bind to p65 mRNA. MiR-370 represses NF-κB activation and inhibits the migration and invasion of HCC cells through downregulation of Lin28A. Sun et al.211 confirmed the tumor suppressor role of miR-370 in HCC cells, showing that miR-370 inhibited HCC cell proliferation and colony formation ability by activating the PI3K/Akt signaling pathway.
In addition to miRNAs that inhibit inflammatory signaling, some miRNAs, such as miR-155, miR-362, miR-301a, and miR-1180, promote the inflammatory response by repressing negative regulators of inflammatory signaling171–173,187. MiR-1180 is upregulated in HCC tissues and HCC-derived cell lines. The NF-κB signaling pathway is activated by miR-1180 overexpression, which occurs through the downregulation of miR-1180 direct targets, OTUD7B and TNIP2. Another study confirmed the expression of TNIP2, a direct target of miR-1180, in HCC cells212. The ectopic expression of miR-1180 promoted the proliferation of HCC cells and inhibited apoptosis because NF-κB induces the expression of genes that participate in pro-survival and anti-apoptotic processes. Ni et al.173 demonstrated that miR-362-5p, which is frequently upregulated in HCC, promoted NF-κB signaling by downregulating CYLD; CYLD is a protein that binds to NF-κB essential modulator and functions as a negative regulator of NF-κB signaling213.
MiRNAs as mediators of inflammatory signaling in HCC
Inflammatory signaling pathway activation was associated in hepatocarcinogenesis. These inflammatory pathways and related transcription factors can affect the development of HCC by regulating miRNA expression. Several studies confirmed that inflammatory signaling facilitates the development of HCC through the downregulation or upregulation of miRNAs in cells (Table 3).
MiRNAs as mediators of inflammatory signaling in HCC
MiR-21
Ning et al.198 observed feedback regulation of HNF4α and NF-κB in HCC. Their study revealed that HNF4α dramatically suppresses the NF-κB signaling pathway and that RelA overexpression or knockdown decreases or increases the expression of HNF4α, respectively. During chronic hepatic inflammation, NF-κB induced the expression of miR-21, which represses HNF4α expression and contributes to hepatocyte de-differentiation and hepatocarcinogenesis. In addition to direct induction of miR-21 expression by NF-κB, miR-21 transcriptional activity is enhanced by high-mobility group box-1 protein (HMGB1), an important participant in inflammation. HMGB1 is a conserved nuclear protein that functions as a chromatin binding factor and allows transcription factors to access specific DNA binding sites219. HMGB1 enhances p65/p50 or p50/p50 binding to specific DNA sites rather than the binding of p65/p65, c-Rel/c-Rel, p65/c-Rel, and p50/c-Rel220. Given its role in the inflammatory cascade, HMGB1 is upregulated in HCC and induces miR-21 expression in HCC cells. The HMGB1/miR-21 signaling axis inhibits the downstream targets RECK and TIMP3 as well as promotes cell proliferation, invasion, and migration221.
MiR-143
MiR-143 is upregulated in HCC tissues, indicating that miR-143 serves as a tumor promoter. MiR-143 expression was increased by HBx-mediated NF-κB activation, although metastasis will be suppressed by miR-143 inhibition214. NF-κB activates transcription by specifically binding to the putative miR-143 promoter, as the NF-κB binding site identifies 5.06 kb upstream of miR-143. MiR-143 promotes cell invasion and tumor metastasis by suppressing the direct target gene FNDC3B. Moreover, miR-143 inhibition results in apoptosis of HCC cells and suppression of HCC tumorigenesis222. In addition, miR-143 was upregulated in serum samples from patients with chronic hepatitis and HCC and therefore might be a useful biomarker for both diseases223.
MiR-221
In addition to the upregulation of miR-221 by HBV, miR-221 can also be induced by HCV infection in an NF-κB-dependent manner99. MiR-221 and NF-κB expression are upregulated after HCV infection. However, the increased expression of miR-221 was blocked after treatment with PDTC, an NF-κB inhibitor. This finding suggests that the HCV-induced upregulation of miR-221 requires NF-κB activation and that NF-κB signaling pathway plays an important role in inflammation-associated cancer.
MiR-224
The expression of miR-224 was upregulated in both cirrhotic livers and HCC samples, suggesting the use of miR-224 as a biomarker of liver inflammation and HCC. The upregulation of miR-224 begins in the pre-cancerous stage and continues during HCC development224. In one study, patients with lower miR-224 expression exhibited a favorable trend of survival. In addition, miR-224 levels were significantly higher in patients with advanced HCC stages than in patients with early stages. MiR-224 could be induced by a set of pro-inflammatory factors, such as TNFα, LPS, and LTα, suggesting that NF-κB signaling regulates miR-224. NF-κB binds to the regulatory region of miR-224 and upregulates miR-224 expression215. Inflammation-mediated activation of miR-224 promotes HCC cell migration and invasion by targeting Rho GTPase-activating proteins. As an onco-miR, the overexpression of miR-224 also facilitates the migration and invasion of HCC cells by targeting HOXD10 and promotes proliferation by targeting p21225,226.
MiR-197
MiR-197 is well-studied during the progression of various cancers in which it plays a role in apoptosis, proliferation, angiogenesis, metastasis, and drug resistance. A negative correlation between miR-197 expression and IL-6 and STAT3 protein levels was observed in HCC tissues, indicating that miR-197 is involved in the STAT3 signaling pathway182. IL-6 is a well-known pro-inflammatory factor that is upregulated during an inflammatory state227. The expression of mature miR-197 was reduced by IL-6 stimulation, whereas the expression of pri-miR-197 remained unchanged by IL-6 stimulation in HCC cells. Further investigation revealed that Drosha binding to pri-miR-197 decreased after IL-6 stimulation, indicating that IL-6 downregulated miR-197 in a post-transcriptional manner by inhibiting Drosha binding to pri-miR-197. Moreover, STAT3 was identified as a direct target of miR-197. As a tumor suppressor, miR-197 inhibits proliferation and invasiveness of HCC cells as well as suppresses the tumorigenesis of HCC xenografts182.
MiR-23a
The first evidence of the role of miR-23a in HCC development was revealed in 2008. MiR-23a induced by TGFβ is remarkably increased in HCC tissues and functions as an anti-apoptotic and pro-proliferative factor in HCC cells228. Later on, Wang et al.218 found that miR-23 can be induced by IL-6 and STAT3 signaling, as a potential STAT3 binding site was identified in the miR-23a promoter region. STAT3-activated miR-23a downregulates the target genes G6PC and PCG-aα and suppresses gluconeogenesis; this reduction in gluconeogenesis facilitates HCC tumorigenesis.
MiRNAs as therapeutic targets in inflammation-related HCC
MiRNAs have a well-recognized role in liver inflammation and hepatocarcinogenesis; many miRNAs are dysregulated during chronic liver inflammation and HCC progression. miRNAs were regarded as novel targets for HCC therapy; the first study using miRNA-based therapy was conducted in 2005. In that study, miR-122 expression was silenced by antagomirs, which act as efficient and specific silencers of endogenous miRNAs. One particular antagomir was injected into mice and dramatically reduced endogenous miR-122 levels229. MiR-122 is a liver-enriched miRNA that functions as a tumor suppressor. A clinical trial of treating HCV infection by miR-122 inhibition was performed because miR-122 targets HCV genomic RNA and promotes HCV viral translation. Jong-Kook Park and colleagues treated tumor-bearing mice with chol-anti-miR-221 or a cholesterol-labeled control oligo. Consistent with the in vitro results, improved survival was observed in the group of mice treated with chol-anti-miR-221158.
In addition, miR-21 is a tumor promoter and functions as a prognostic factor, as previously described. MiR-21 promotes the proliferation, metastasis, and tumorigenesis of HCC and was validated as an onco-miR involved in drug resistance. MiR-21 suppression by lenti-miR-21-i combined with 5-fluorouracil and pirarubicin treatment leads to significant suppression of HCC cell proliferation; 5-fluorouracil and pirarubicin downregulated the miR-21 expression through AP-1 signaling230.
Oncolytic viruses were proposed as a novel therapeutic strategy for cancer treatment. Ad5-based oncolytic viruses were proven effective and safe in clinical trials231. MiR-199 is significantly reduced in HCC tissues compared with normal tissues. MiR-199 was cloned into adenoviral vectors to produce miR-199 adenovirus. Tumor-bearing nude mice were intra-tumorally injected with either miR-199 adenovirus or PBS as a control. Tumor growth was dramatically suppressed in the group of mice treated with miR-199 adenovirus. These results were confirmed in a transgenic model in which mice were predisposed to HCC development. The group of mice treated with miR-199 adenovirus exhibited a reduced tumor burden and a decreased number of tumor nodules compared with the control group. It can be expected in the future that any miRNAs acting as tumor suppressors can be applied as therapeutic oncolytic viruses in a manner similar to miR-199232.
Another miRNA-based therapeutic strategy is the design of miRNA panels that targets a particular protein to modulate inflammatory response and HCC development. For example, repressor/activator protein (Rap) might protect telomeres. Rap1 is an integral component of the IκB kinase (IKK) complex. The association between the IKK complex and Rap1 is important for NF-κB activation because Rap1 serves as an adaptor to recruit IKKs and aids in phosphorylation of the NF-κB p65 subunit233. Rap1 knockdown sensitized cancer cells to chemotherapeutic drug-induced apoptosis. 5-fluorouracil is a widely used chemotherapy drug for advanced HCC patients, but many patients with HCC are highly resistant to this chemotherapy regimen. A set of 4 Rap1-miRNAs was designed to knock down Rap1, leading to a significant increase in the sensitivity of HepG2 cells to 5-fluorouracil-induced apoptosis234.
A number of miRNAs can be induced or inhibited during hepatitis because of viral or non-viral infections. Some miRNAs are also activated or repressed when inflammatory signaling pathways, such as NF-κB and JAK/STAT3 signaling, cannot be turned off. In turn, miRNAs that might function as oncogenes or tumor suppressors can promote or suppress both inflammatory cascades in hepatocarcinogenesis. This function indicates that miRNAs are novel regulators or mediators of liver inflammation and hepatocarcinogenesis (Figure 1). Tumor tissues can be highly complex and heterogeneous. The tumor microenvironment consists of many types of cells including tumor invading immune-cells, fibroblasts, vascular endothelial cells, lymphatic cells, and stromal components. Given that numerous components of tumor microenvironment are responsible for the initiation, development, and progression of human cancer235, the dysregulated miRNAs from tumor specimens might originate from different types of cells in the tumor microenvironment and function in human carcinogenesis by multiple ways. The next investigations should focus on the origin, location, action, and multi-function of the dysregulated miRNAs in initiation, development, and progression of HCC. Additionally, any study of cell lines might run the risk of false discovery. The results from cell lines cannot reflect the full aspects of the pathological process of HCC. The next investigations on the exact role of miRNAs in inflammation-associated HCC should be verified in patients and reliable HCC models.
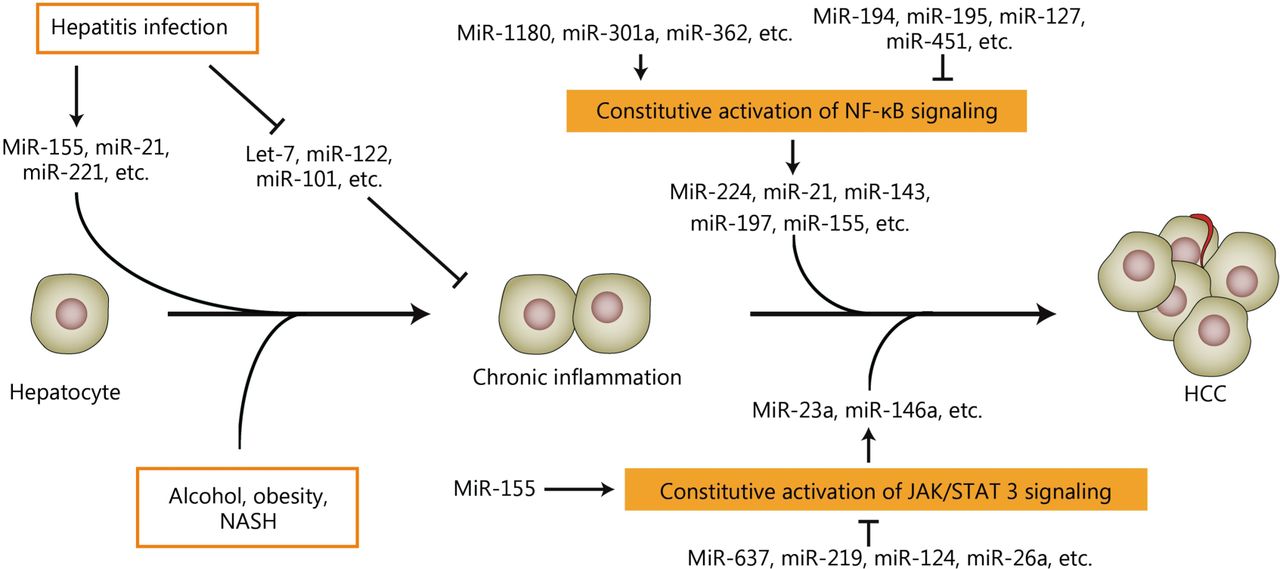
A simplified sketch illustrates the development of liver inflammation and HCC and shows the key nodes in which aberrantly expressed miRNAs participate in inflammation-associated hepatocarcinogenesis. Many miRNAs are aberrantly expressed when liver inflammation occurs and play prominent roles in HCC development. MiRNAs might be regulated by constitutive inflammatory signaling pathways and participate in the development and progression of HCC. Additionally, miRNAs might modulate the inflammatory signaling pathways during the development and progression of HCC.
The 5-year overall survival of HCC is lower than 10% worldwide because many patients are diagnosed at advanced stages236. Therefore, discovering novel viable biomarkers for HCC early diagnosis is urgent. In the last decades, numerous pieces of evidence highlighted the regulatory roles of miRNAs in inflammation-associated HCC. Evidence also indicated that the dysregulated miRNAs might be useful as biomarkers for liver inflammation and HCC progression. In 2015, miRNAs were screened in a large-scale, multicenter, retrospective longitudinal study to explore effective and reliable strategies for monitoring at-risk populations that suffers from chronic inflammation as well as detect HCC at an early stage. Serum miR-29a, miR-29c, miR-133a, miR-143, miR-145, miR-192, and miR-505 might act as biomarkers in HCC because they are in significantly higher content in HCC compared with healthy controls or those with chronic inflammation. The classifier composed of these seven miRNAs could be more sensitive than α-fetoprotein in distinguishing patients with HCC from those who are cancer-free237. The distinction requires large-scale, multicenter, independent investigations to confirm its feasibility. Several studies illustrated the potential of miRNAs as therapeutic targets to prevent or treat HCC. However, additional profiles and detailed mechanisms of miRNA functions are required to provide a better understanding of their role in liver inflammation and hepatocarcinogenesis. The additional data will facilitate the application of more miRNAs into prevention and therapy for inflammation-associated HCC.
Conflict of interest statement
No potential conflicts of interest are disclosed.
Acknowledgments
This work was supported by grants from the National Key Basic Research Program (Grant No. 2013CB910504), and the National Natural Science Foundation of China (Grant No. 81125016 and 81472565).
- Received August 24, 2016.
- Accepted September 18, 2016.
- Copyright: © 2016, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.
- 50.
- 51.↵
- 52.↵
- 53.
- 54.
- 55.
- 56.
- 57.
- 58.
- 59.
- 60.
- 61.
- 62.
- 63.
- 64.
- 65.
- 66.
- 67.
- 68.
- 69.
- 70.
- 71.
- 72.
- 73.
- 74.
- 75.
- 76.
- 77.
- 78.
- 79.
- 80.
- 81.
- 82.
- 83.
- 84.
- 85.
- 86.
- 87.
- 88.
- 89.
- 90.
- 91.
- 92.
- 93.
- 94.
- 95.
- 96.
- 97.
- 98.↵
- 99.↵
- 100.
- 101.
- 102.
- 103.
- 104.↵
- 105.
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.
- 140.
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.
- 154.
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.
- 162.↵
- 163.↵
- 164.↵
- 165.
- 166.
- 167.↵
- 168.
- 169.
- 170.
- 171.↵
- 172.
- 173.↵
- 174.
- 175.
- 176.↵
- 177.↵
- 178.
- 179.↵
- 180.↵
- 181.
- 182.↵
- 183.↵
- 184.↵
- 185.
- 186.↵
- 187.↵
- 188.↵
- 189.↵
- 190.↵
- 191.↵
- 192.↵
- 193.↵
- 194.
- 195.
- 196.↵
- 197.↵
- 198.↵
- 199.↵
- 200.↵
- 201.↵
- 202.↵
- 203.↵
- 204.↵
- 205.↵
- 206.↵
- 207.
- 208.
- 209.
- 210.↵
- 211.↵
- 212.↵
- 213.↵
- 214.↵
- 215.↵
- 216.
- 217.
- 218.↵
- 219.↵
- 220.↵
- 221.↵
- 222.↵
- 223.↵
- 224.↵
- 225.↵
- 226.↵
- 227.↵
- 228.↵
- 229.↵
- 230.↵
- 231.↵
- 232.↵
- 233.↵
- 234.↵
- 235.↵
- 236.↵
- 237.↵
In this issue






{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.