

Abstract
Objective: Hepatocellular carcinoma (HCC) is a leading cause of cancer-related deaths. Novel serum biomarkers are required to increase the sensitivity and specificity of serum screening for early HCC diagnosis. This study employed a quantitative proteomic strategy to analyze the differential expression of serum glycoproteins between HCC and normal control serum samples.
Methods: Lectin affinity chromatography (LAC) was used to enrich glycoproteins from the serum samples. Quantitative mass spectrometric analysis combined with stable isotope dimethyl labeling and 2D liquid chromatography (LC) separations were performed to examine the differential levels of the detected proteins between HCC and control serum samples. Western blot was used to analyze the differential expression levels of the three serum proteins.
Results: A total of 2,280 protein groups were identified in the serum samples from HCC patients by using the 2D LC-MS/MS method. Up to 36 proteins were up-regulated in the HCC serum, whereas 19 proteins were down-regulated. Three differential glycoproteins, namely, fibrinogen gamma chain (FGG), FOS-like antigen 2 (FOSL2), and α-1, 6-mannosylglycoprotein 6-β-N-acetylglucosaminyltransferase B (MGAT5B) were validated by Western blot. All these three proteins were up-regulated in the HCC serum samples.
Conclusion: A quantitative glycoproteomic method was established and proven useful to determine potential novel biomarkers for HCC.
keywords
Introduction
Hepatocellular carcinoma (HCC) ranks the second leading cause of cancer-related deaths worldwide1. HCC is usually associated with viral infections, such as hepatitis C and hepatitis B. HCC diagnosis is generally based on ultrasound scanning and serum alpha-fetoprotein (AFP) estimation every 6 months2. However, ultrasound can only detect tumors larger than 3 cm, and this method cannot distinguish malignant tumors from benign ones. Other methods such as computed tomography and nuclear magnetic resonance are usually very expensive for routine screening3. Serum is an ideal sample source for HCC detection. The known serum biomarkers for HCC include AFP, des-γ-carboxyprothrombin (DCP), fucosylated AFP (AFP-L3), Golgi protein 73 (GP73), isozyme of alkaline phosphatase (variant ALP), and isozyme of gamma-glutamyl transpeptidase (novel γ-GTP)4,5. AFP is a classical biomarker, and an increase in AFP serum level correlates with tumor size6. However, in patients with chronic liver diseases, AFP concentration is also slightly increased. DCP is an abnormal prothrombin that has been used for clinical HCC screening. High DCP level reflects a poor prognosis7. DCP sensitivity for early and small HCC (<2 cm) is only 56.5%8. AFP-L3 is a fucosylated species of AFP, and the serum concentration of AFP-L3 is associated with poor HCC differentiation9. Novel serum biomarkers are urgently required to increase the sensitivity and specificity of serum biomarker screening for early HCC diagnosis.
Mass spectrometry-based proteomic approaches have evolved as powerful tools to discover novel biomarkers10,11. However, identification of potential protein biomarkers from biofluid samples, such as serum and plasma, remains challenging because of their large protein concentration range. Efforts have been made to simplify serum samples via affinity chromatography, either by removing abundant proteins from the serum or enriching a subproteome with a common chemical structural feature12-14, e.g., affinity depletion using antibody-conjugated materials12,13. The interest in abnormal protein glycosylation research is increasing. The currently known HCC biomarkers, namely, AFP, AFP-L3, DCP, and GP73, are all glycoproteins. Many biomarkers clinically used for cancer diagnosis are also glycoproteins, such as prostate-specific antigen in prostate cancer15; Her2/neu in breast cancer16; CA-125 in ovarian cancer17; and CEA in colorectal, breast, pancreatic, and lung cancer18. Therefore, targeting glycoproteins in the serum can enrich the potential biomarkers while reducing the serum sample complexity for in-depth proteome analysis.
In this study, lectin affinity chromatography (LAC) was used to enrich glycoproteins from blood samples of 40 healthy volunteers and 40 HCC patients. Stable isotope dimethyl labeling and 2D liquid chromatography (LC) separation were used for quantitative mass spectrometric analysis to examine the differential levels of the detected proteins. More than 2,000 proteins were characterized in the serum. A panel of proteins exhibited significant changes in relative abundances between the HCC and control samples. The expression patterns of fibrinogen gamma chain (FGG), FOS-like antigen 2 (FOSL2), and α-1,6-mannosylglycoprotein 6-β-N-acetylglucosaminyltransferase B (MGAT5B) were validated by Western blot.
Materials and methods
Serum collection
The study was approved by the Ethics Committee of Tianjin Medical University. Serum samples were processed from each individual by using a 12G BD Vacutainer Safety-LokTM blood collection system. After collection, samples were immediately placed on ice and allowed to stand for 30 min. Samples were then centrifuged at 3,000 rpm for 15 min and stored at −80 °C until analysis. A total of 40 HCC serum samples were collected and divided into two groups. Each serum cohort, which consisted of 20 HCC samples and 20 cases of age- and gender-matched normal control cohort, was pooled for quantitative glycoproteomic analysis. HCC diagnoses were confirmed through histopathologic study.
Reagents and materials
Iodoacetamide, N-acetyl-D-glucosamine, methyl-R-D-mannopyranoside, methyl-R-D-glucopyranoside, manganese chloride tetrahydrate, formaldehyde, deuterated formaldehyde, and sodium cyanoborohydride were purchased from Sigma-Aldrich (St. Louis, MO). Agarose-bound Concanavalin A (Con A, 6 mg lectin/mL gel) and wheat germ agglutinin (WGA, 7 mg lectin/mL gel) were purchased from Vector Laboratories (Burlingame, CA). Dithiothreitol (DTT) and sequencing grade modified trypsin were purchased from Promega (Madison, WI). Antibodies used in Western blot were all obtained from Santa Cruz Biotechnology (Dallas, TX).
Lectin affinity chromatography (LAC)
Lectin affinity columns were prepared by adding 400 μL each of Con A and WGA slurry to empty Micro Bio-Spin columns (Bio-Rad Laboratories, Hercules, CA), as reported by Wei et al.19. Con A exhibited a high affinity to high-mannose type N-glycans, whereas WGA was selective for N-acetyl-glucosamine (GlcNAc). Up to 40 mL of pooled serum was diluted 10 times with the binding buffer (20 mM Tris, 0.15 M NaCl, 1 mM CaCl2, 1 mM MnCl2, pH 7.4) and loaded onto the lectin affinity columns. After shaking for 6 h, unretained proteins were discarded, and lectin beads were washed with 2.5 mL of binding buffer. The captured glycoproteins were eluted with 2 mL of elution buffer (10 mM Tris, 0.075 M NaCl, 0.25 M Nacetyl-D-glucosamine, 0.17 M methyl-R-D-mannopyranoside, and 0.17 M methyl-R-D-glucopyranoside). The eluted fraction was concentrated using a 10 kDa Centricon Ultracel YM-10 filter (Millipore, Billerica, MA). BCA assays were performed to measure the protein concentration.
Protein digest and stable isotopic labeling
Concentrated samples were denatured with 6 M urea in 0.2 M sodium acetate buffer (pH 8) and reduced by incubation with 10 mM DTT at 37 °C for 1 h. Reduced proteins were alkylated for 1 h in darkness with 40 mM iodoacetamide. Alkylation reaction was quenched by adding DTT to a final concentration of 50 mM. Samples were diluted to a final concentration of 1 M urea. Trypsin was added at a 50:1 protein-to-trypsin mass ratio, and samples were incubated at 37 °C overnight. Sodium cyanoborohydride was added to the protein digest to a final concentration of 50 mM. Samples were labeled with either 0.2 mM formaldehyde or 0.2 mM deuterated-formaldehyde. The mixed peptides were vortexed and incubated at 37 °C for 1 h. Up to 2 M NH4OH was added to quench the reaction, and the mixture was immediately dried using SpeedVac. Finally, samples were reconstituted in water.
High-pH reversed-phase liquid chromatography (RPLC)
Equal amounts of light- and heavy-labeled samples were combined and separated using Waters HPLC C18 columns with high pH stability at a flow rate of 150 μL/min. The peptides were eluted with a 40 min gradient 5%-45% buffer B (buffer A: 100 mM ammonium formate, pH 10; buffer B: acetonitrile). Fractions were collected every 3 min for 60 min. Collected fractions were dried using SpeedVac and reconstituted in 20 μL of 0.1% formic acid. Up to 2 μL of each of the 10 fractions containing peptides was subjected to LC-MS/MS.
LC-MS/MS and data analysis
A nanoUPLC system (Waters, Milford, MA) was used to separate the tryptic peptides. Samples were loaded on a C18 trap column and flushed with mobile phase A (0.1% formic acid in H2O) at 5 μL/min for 10 min before being delivered to a nanoUPLC column (C18, 150 mm × 0.075 mm × 1.7 μm). The peptides were eluted using a 7%-45% B gradient (0.1% formic acid in acetonitrile) over 90 min into a nano-electrospray ionization (nESI) LTQ Orbitrap mass spectrometer (ThermoFisher Scientific, Waltham, MA). The mass spectrometer was operated in data-dependent mode, in which an initial FT scan recorded the mass range of m/z 350-2,000. The spray voltage was set between 1.8 and 2.0 kV, and the mass resolution used for MS scan was 30,000. For tandem mass analysis, the eight most abundant ions were automatically selected for collisionally activated dissociation. The mass window for precursor ion selection was m/z ±1, and the normalized collision energy was set at 35% for MS/MS. Dynamic exclusion parameters were set as follows: exclusion duration was 60 s, and exclusion mass width was 0.01% relative to reference mass.
Raw data were searched through UniProt human protein database containing 98,778 sequence entries via SEQUEST algorithm embedded in the Protein Discoverer version 1.3 software (ThermoFisher Scientific, Waltham, MA). The following parameters were applied during the database search: 10 ppm precursor mass error tolerance, 1 Da fragment mass error tolerance, static modifications of carbamidomethylation for all cysteine residues, dimethylation for formaldehyde-labeled sample (+28 Da) or deuterated-formaldehyde-labeled (+32 Da) lysine, and N-terminus. False discovery rate of <0.05 was used as filtering criteria for all identified peptides. Proteins identified with the same set of peptides were grouped and treated as one. Protein Discoverer was used for relative quantification. Two groups of pooled serum samples were analyzed, each of which contained serum collected from 20 patients or healthy donors. Each sample was analyzed thrice. Protein identification information was imported to PANTHER database for gene ontology analysis (http://www.pantherdb.org).
Results
Enrichment of glycoproteins from serum
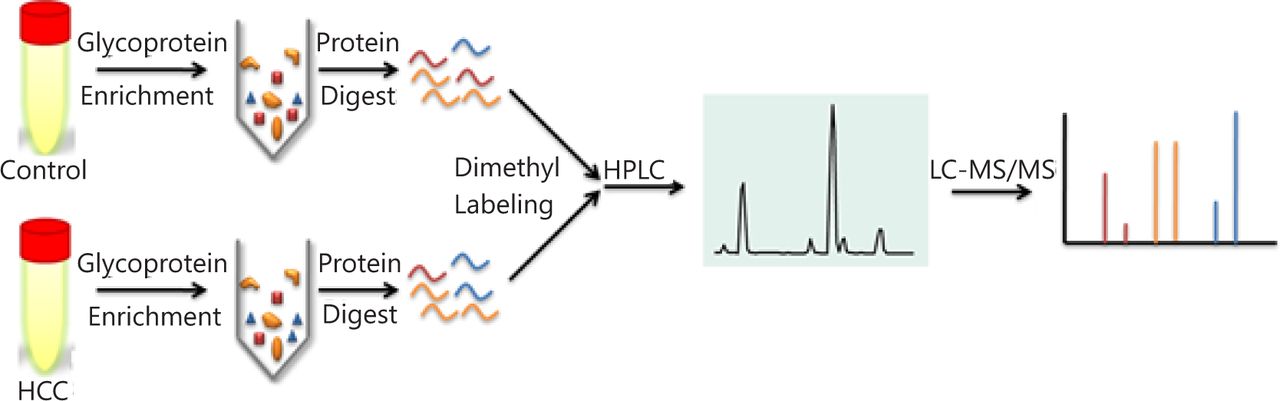
Direct proteome analysis in serum was challenging because of the presence of highly abundant proteins. The analyzing strategy employed in this study is shown in Figure 1. A lectin affinity column with a broad sugar selection spectrum was used to purify the glycoproteins from the serum. Two lectin types were combined to extend the coverage of glycoprotein enrichment. Con A showed a high affinity to high-mannose type N-glycans, whereas WGA was selective for GlcNAc. Majority of the serum albumin and other abundant proteins were removed from the serum, which reduced the sample complexity and increased the detection sensitivity for proteins with low abundance. Approximately 80% of the total serum proteins were removed after LAC purification (Figure 2A). Glycoprotein enrichment effectiveness was further demonstrated by the mass spectrometry results. With lectin selection, albumin no longer appeared on the top of the protein identification list, although a moderate number of albumin peptides can still be detected probably because of nonspecific and secondary binding events.

Quantitative proteomic schematic of serum glycoproteins. Glycoproteins in serum samples were enriched using lectin affinity chromatography and digested by trypsin then labeled using isotopic formaldehyde, followed by 2D RP-RP LC-MS/MS.
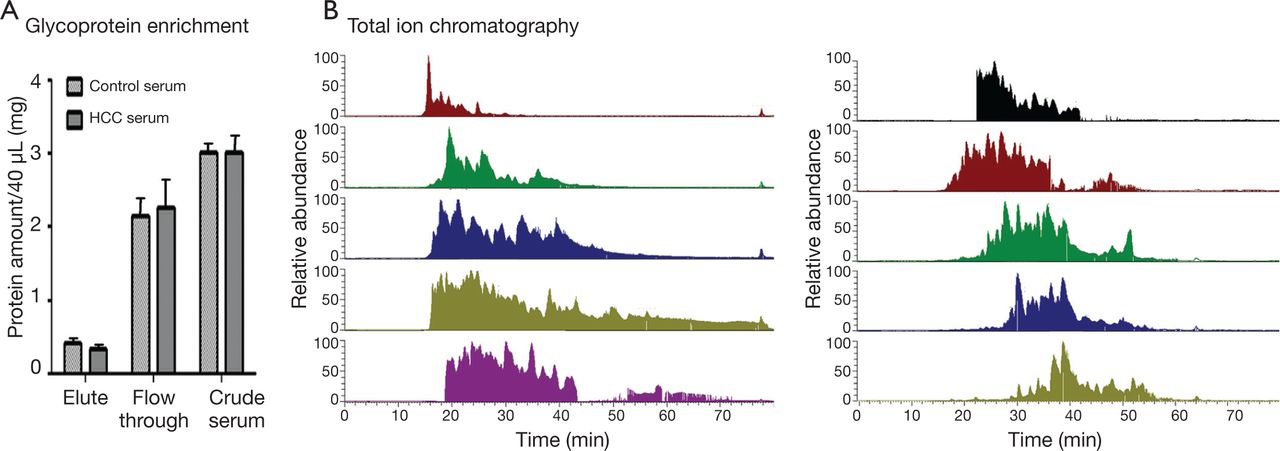
Sample serum glycoprotein preparation. (A) The protein content in the crude serum, flow-through from LAC column, and the eluted fraction that contained glycoproteins, as measured using BCA assay. (B) LC-MS/MS total ion chromatogram from 10 fractions separated by high pH RP-LC separation.
Relative glycoprotein quantification between HCC and control serum
A total of 40 HCC and control samples were analyzed to investigate the differential serum glycoprotein expression induced by HCC. Patient information is provided in Table 1. Multidimensional separations were extensively applied in proteomic studies to reduce the complexity of samples. In the present study, RPLC was used in both dimensions of the separation under significantly different pH conditions. Moreover, 2D RP-RP LC-MS/MS demonstrated great orthogonality in peptide separations because of charge changes in acidic and basic amino acid side chains under different pH conditions20. The use of RP as first dimension provided higher separation resolution and higher peptide recovery than strong cation exchange. Different peptide total ion chromatogram profiles were observed between the fractions collected from the first dimension of RPLC separation (Figure 2B). The widespread of peaks in all 10 fractions in the second dimension provided great resolution to enhance proteomic detection.
Clinical information of HCC samples and normal controls
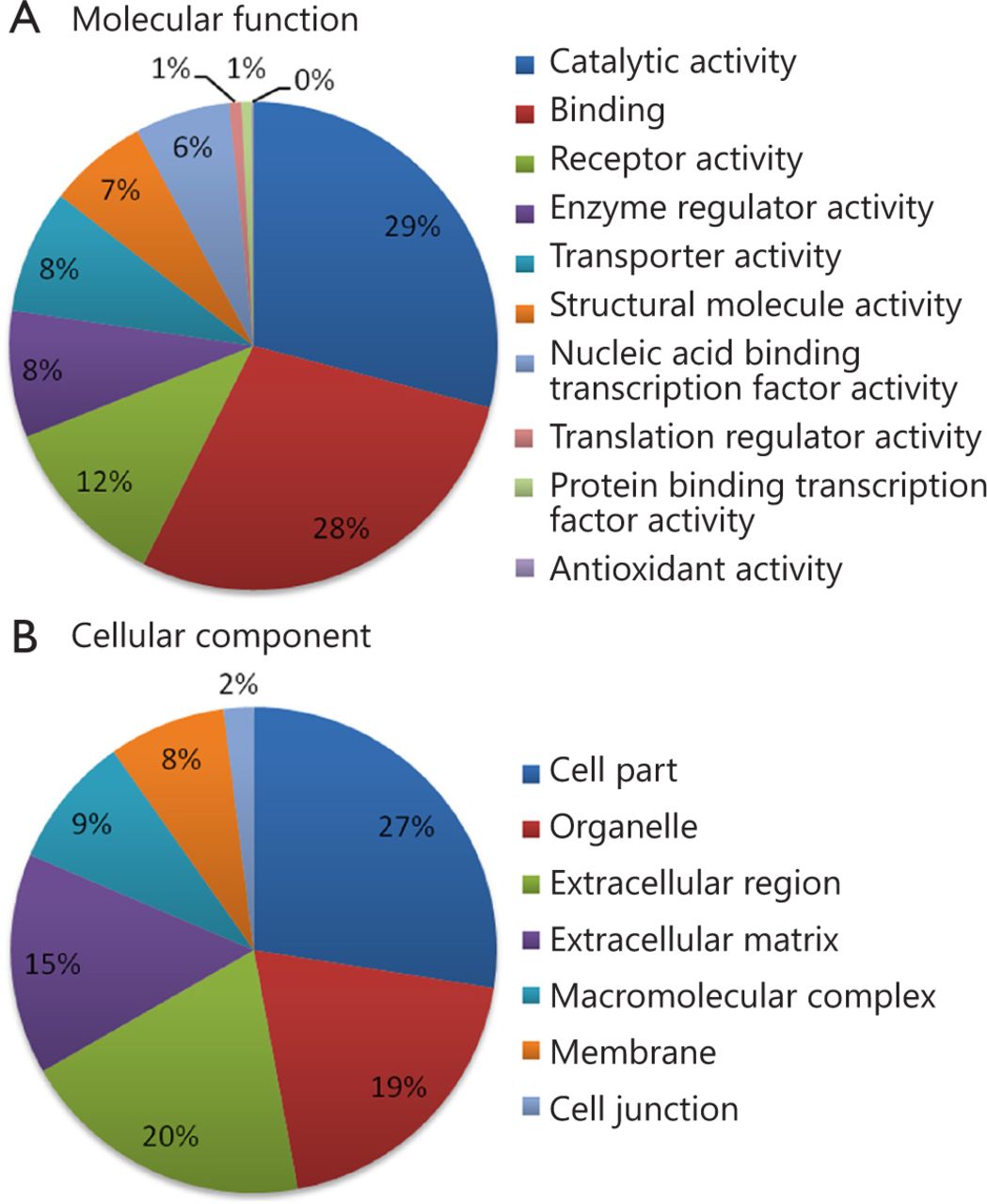
Using 2D RP-RP LC-MS/MS method, 2,280 protein groups were identified from the two groups of pooled serum samples (Table S1 in the supplementary materials, available with the full text of this article at www.cancerbiomed.org). Systematic gene ontology analysis was performed using PANTHER database. Molecular function analysis revealed that the majority of the identified serum proteins demonstrated catalytic (29%), binding (28%), and receptor activities (12%) (Figure 3). Cellular component analysis showed that most of the identified serum proteins probably originated from tissue leakage, including cell parts (27%) and organelles (19%). Another large portion resulted from the extracellular region (20%) and extracellular matrix (15%).
A total of 2,280 protein groups identified from the two groups of pooled serum samples
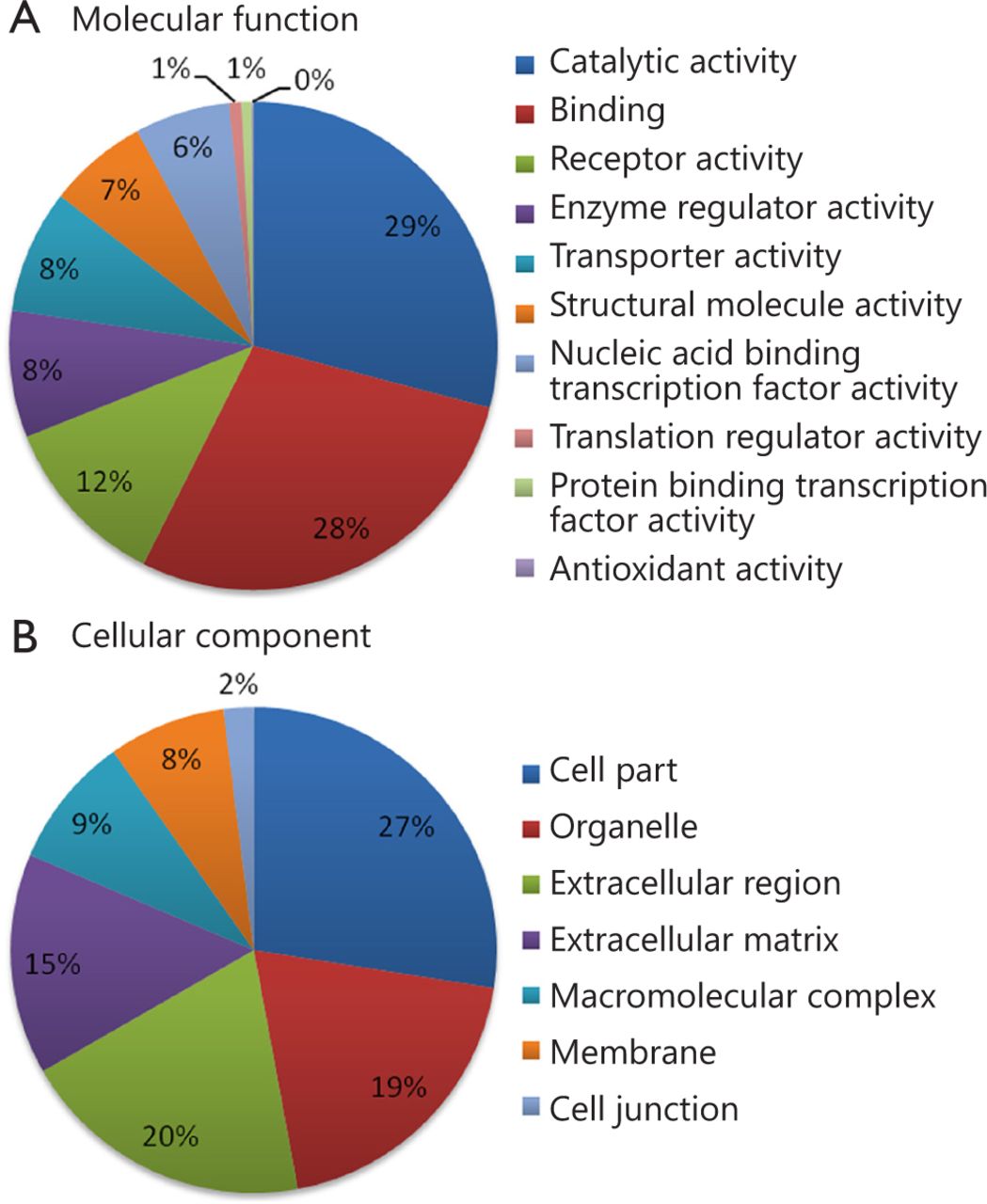
Gene ontology analysis of identified serum proteins. (A) Diagram showing the molecular functions of the identified proteins. (B) Diagram showing the cellular components of the identified proteins. Annotation information was acquired through PANTHER database (http://www.pantherdb.org).
Quantitative proteomic analysis was performed to calculate the ratios of the identified serum glycoproteins between HCC and control. Given the low glycoprotein concentration, proteins identified with only one unique peptide were kept in the list. However, further validation is required. Twofold cutoff threshold showed that 36 proteins were up-regulated in the HCC serum, whereas 19 proteins were down-regulated (Table 2). AFP, the clinically used marker for HCC diagnosis, was detected in HCC serum only.
List of differently expressed glycoproteins in human serum from patients with hepatocellular carcinoma
Validation of three differently expressed proteins
Besides AFP, other glycoproteins with differential expression levels were also found: FGG, FOSL2, and MGAT5B. To validate this observation in quantitative MS experiments, Western blot analysis was performed to test the level of the three up-regulated glycoproteins in seven groups of serum samples. Abundant protein depletion was performed to remove albumin and IgG prior to Western blot analysis. FGG, MGAT5B, and FOSL2 were detected using Western blot (Figure 4). Consistent with the mass spectrometry analysis results, the three proteins were up-regulated in the seven HCC serum samples compared with the normal control. Further studies must be performed on large numbers of clinical samples by using ELISA to evaluate the potential usage of FGG, FOSL2, and MGAT5B as serum biomarkers.

Western blot analysis of identified differential serum proteins. Serum samples were first clarified by depletion of highly abundant proteins. Seven groups of HCC and control serum samples were analyzed. H, HCC; C, control.
Discussion
Novel biomarker discovery using traditional biological assays is time consuming. With the great technical development of mass spectrometry, quantitative proteomics has become an essential tool to determine biomarkers. A large effort has been devoted to mining novel serum HCC biomarkers over the last decade3,21,22. The technology used in these studies evolved from 2D electrophoresis to multidimensional LC-MS/MS. Different analytical methods can provide complementary information, which can validate previous reports and provide new opportunities to discover novel biomarkers. For example, Pan et al.23 reported that two commonly used glycoprotein enrichment methods, namely, LAC and hydrazide coupling enrichment, can purify different glycoprotein pools from serum. Hydrazide coupling and label-free quantification were used in a previous study to examine the glycoprotein expression in HCC serum. In the present study, a different strategy was employed by combining LAC and dimethylation isotopic labeling. Several proteins, such as AFP, fibrinogen beta chain, polymeric-immunoglobulin receptor, and insulin-like growth factor-binding protein 3, were detected and quantified by both LAC and dimethylation isotopic labeling. However, majority of the detected proteins were not reported in previous research.
Three differential glycoproteins were validated using Western blot. FGG role was suggested for other tumor types24,25. Zhu et al.26 demonstrated that FGG was significantly up-regulated at the mRNA level in the HCC cell lines and HCC tissues. Plasma fibrinogen progressively increased with the tumor clinical stage of HCC patients26,27. The higher FGG serum concentration in HCC patients than in healthy people may be attributed to the higher FGG expression in HCC and increased fibrinogen degradation28.
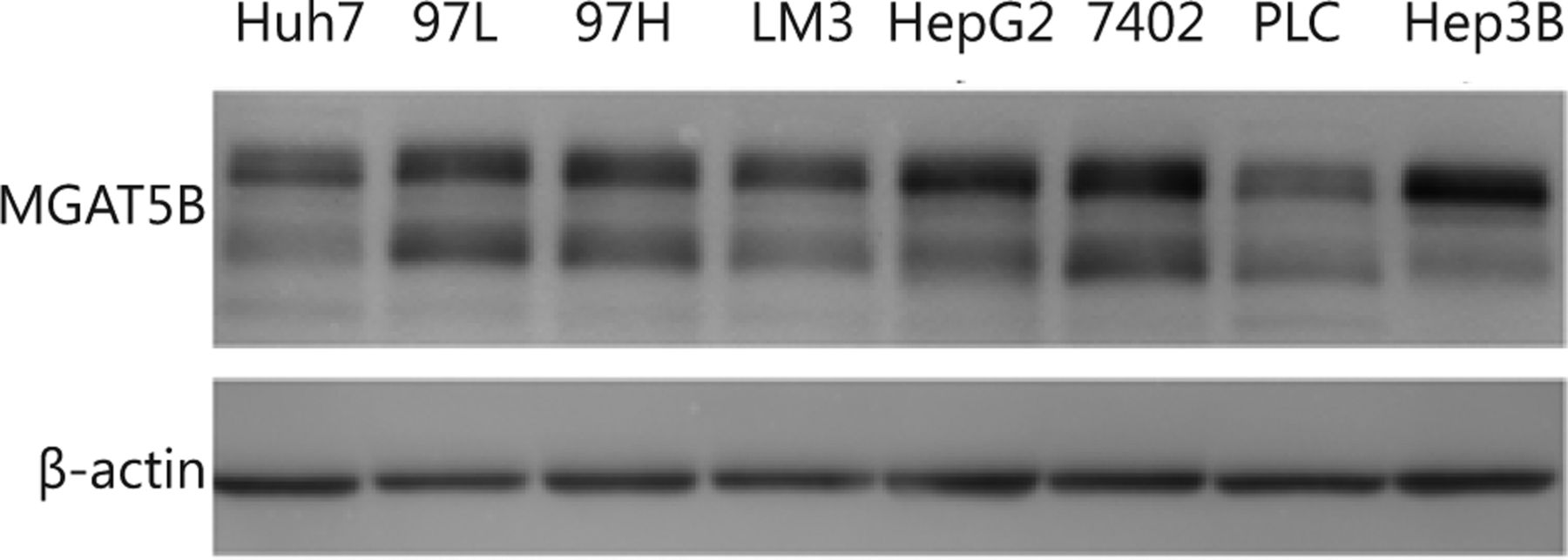
MGAT5B (also reported as GnT-Vb or GnT-IX), an MGAT5 isozyme, demonstrated a broad transfer activity toward GlcNAc β 1,2-Man α 1-Ser/Thr, which formed a 2,6-branched structure in brain O-mannosyl glycan29. MGAT5B was exclusively detected in neural tissues and testes30,31. Lange et al.32 detected an MGAT5B expression in prostate cancer cells and xenografts, whereas MGAT5B was absent in the primary prostate epithelial cells and normal human prostate. Liu et al.33 observed that MGAT5B was up-regulated in metastatic HCC clinical cancer specimens, and the trend was the same in human HCC cell lines and orthotopic xenograft tumors. The current results also revealed increased MGAT5B levels in the serum of HCC patients compared with normal controls. Additionally, MGAT5B was extensively expressed in different HCC cell lines (Figure S1 in the supplementary materials, available with the full text of this article at www.cancerbiomed.org). More studies must be conducted in the future to understand the functions of MGAT5B in HCC development.

Western blotting analysis of MGAT5B from different HCC cell lines. The same amount of protein was loaded for each cell lystae, and β-actin was used as internal standard.
FOSL2 belongs to the activator protein 1 (Jun and Fos family) transcription factors, which regulate gene expression in cell proliferation, differentiation, inflammation, and malignant transformation34. FOSL2 overexpression was associated with the progression of various human tumor types, including breast cancer35, ovarian carcinoma36, salivary gland tumors37, colorectal cancer36-38, and adult T-cell leukaemia39. FOSL2 is one of the genes affected by the characteristic t(2;5) translocation in tumor cells40. The present study suggests for the first time that FOSL2 is up-regulated in HCC serum. However, additional studies are needed to confirm the findings and determine whether such up-regulation is caused by FOSL2 overexpression in HCC tumor tissues.
Supplementary materials
This has been provided by the authors to give readers additional information about their work.
Supplement to: Gao HJ, Chen YJ, Zuo D, Xiao MM, Li Y, Guo H, Zhang N, Chen RB. Quantitative proteomic analysis for high-throughput screening of differential glycoproteins in hepatocellular carcinoma serum. Cancer Biol Med 2015;12:246-254. doi: 10.7497/j.issn.2095-3941.2015.0010
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 21205088), 973 Project (Grant No. 2011CB933100), National Science Fund for Distinguished Young Scholars (Grant No. 81125019), and Doctoral Research Fund from the Ministry of Education of China (Grant No. 20121202120001). This project was also sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
Footnotes
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received January 30, 2015.
- Accepted May 8, 2015.
- Copyright: © 2015, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.