

Abstract
Objective: Glioblastoma is the most common primary brain tumor in adults and one of the most lethal human tumors. It constitutes a unique non-metastasizing human tumor model with high resistance to radiotherapy and chemotherapy. The current study investigates the association between autophagic flux and glioblastoma cell resistance.
Methods: The expression kinetics of autophagy- and lysosome-related proteins following exposure of two glioblastoma cell lines (T98 and U87) to clinically relevant radiation doses was examined. Then, the response of cells resistant to radiotherapy and chemotherapy was investigated after silencing of LC3A, LC3B, and TFEB genes in vitro and in vivo.
Results: Following irradiation with 4 Gy, the relatively radioresistant T98 cells exhibited enhanced autophagic flux. The more radiosensitive U87 cell line suffered a blockage of autophagic flux. Silencing of LC3A, LC3B, and TFEB genes in vitro, significantly sensitized cells to radiotherapy and temozolomide (U87: P < 0.01 and < 0.05, respectively; T98: P < 0.01 and < 0.01, respectively). Silencing of the LC3A gene sensitized mouse xenografts to radiation.
Conclusions: Autophagy in cancer cells may be a key factor of radio-resistance and chemo-resistance in glioblastoma cells. Blocking autophagy may improve the efficacy of radiochemotherapy for glioblastoma patients.
keywords
Introduction
Glioblastoma is the most common primary brain tumor in adults and one of the most lethal human tumors1 with a median survival less than 6 months for inoperable patients. Due to the local growth patterns of the disease, complete surgical removal is difficult and postoperative radiotherapy is necessary for increasing median survival by 6 months2. DNA damaging agents, like temozolomide, have been successfully applied in clinical practice3. Increasing radiotherapy efficacy is a major key in the fight against glioblastoma.
Macroautophagy (or autophagy for simplicity) is an important cell process that degrades accumulated damaged proteins and cell organelles and recycles cellular components. Autophagy is activated under stressful conditions such as hypoxia, starvation, and acidosis4,5. Damaged material becomes engulfed by double-membraned, autophagosomal vesicles that fuse with lysosomes, allowing the degradation of autophagosomal content by lysosomal enzymes. Autophagy-related protein 8 (Atg8)/Microtubule-associated Protein 1 Light Chain 3 (or LC3) is a ubiquitin-like protein that is essential for autophagosomal membrane formation; this protein has two forms, the soluble LC3-I form (18 kDa), derived from a pro-LC3 30 kDa protein after cleavage by autophagin Atg4, and the membrane-bound LC3-II form (16 kDa) that resides in both the inner and outer autophagosomal membrane4,5. Sequestosome-1(SQSTM1)/p62 is a multifunctional protein that interacts with LC3, a central component of the autophagy machinery, and transports ubiquitinated proteins for degradation by the autophagosome. Following autophagosome-lysosome fusion (autolysosome), p62 is degraded; thus, kinetics of p62 levels is a useful marker of autophagic flux6.
As autolysosomal fusion is essential for autophagic flux and transcription factor EB (TFEB) regulates expression of lysosomal proteins, such as lysosomal-associated membrane proteins (LAMPs) and other lysosomal membrane proteins and enzymes that mediate degradation of autolysosomal content, TFEB thus plays an important role in the autophagic cell response7.
Studies have shown that glioblastoma cells exhibit resistance to apoptosis after exposure to ionizing radiation and that autophagic cell death is the prevalent pathway involved in glioblastoma tumor radiation response8. Modulation of apoptosis-related genes, such as phosphatase and tensin homolog (PTEN), may be involved in this phenomenon9. Ito et al.10 found that ionizing radiation induces cell cycle arrest and autophagic cell death—but not apoptosis—in glioma cell lines. Therefore, overcoming glioblastoma cell resistance to autophagic cell death is a potential therapeutic strategy against glioblastoma that demands thorough investigation8,11.
In the current study, we investigated the expression kinetics of autophagy- and lysosome-related proteins following the exposure of two glioblastoma cell lines, T98 and U87 (the latter lacking PTEN expression), to clinically relevant radiation doses. The radiosensitizing effect of silencing the expression of key autophagy (LC3A and LC3B) and lysosomal biogenesis (TFEB) molecules was also studied.
Materials and methods
Cell lines and cultures
U87MG and T98G were purchased from American Type Culture Collection (ATCC® HTB-14TM and ATCC® CRL-1690TM, respectively, Manassas, USA). Details regarding the cell lines can be found at https://www.lgcstandards-atcc.org/Products/All/HTB-14.aspx and https://www.lgcstandards-atcc.org/Products/All/CRL-1690.aspx. Cell line authentication was performed by ATCC and details regarding characterization methodology are available at http://www.atcc.org/Products/Cells_and_Microorganisms/Testing_and_Characterization/STR_Profiling_Analysis.aspx.
Cells were cultured under aseptic conditions using Dulbecco’s Modified Eagle Medium (DMEM) (31885-023; Gibco, MD, USA) supplemented with 10% fetal bovine serum (FB-1000/500; Biosera, London, UK), 100 units/mL penicillin-streptomycin (15140-122; Gibco, MD, USA) and 2 mM L-glutamine (25030; Gibco). Both cell lines were maintained at standard conditions (37°C and 5% CO2) in a humidified atmosphere and were harvested upon reaching 70%–90% confluency.
Small interfering RNA (siRNA) transfection assays
siRNA sequences targeting LC3A, LC3B, and TFEB were designed by and purchased from GenePharma (Shanghai, China). A pool of 4 specific sequences were used for LC3A and TFEB genes as follows, 5′-GCGAGUUGGUCAAGAUCAUTT-3′, 5′-GCUUCCUCUAUAUGGUCUATT-3′, 5′-CCUGCUGUGUGGUUCAUCUTT-3′, and 5′-GCUGUAAGGAGGUACAGCATT-3′, TFEB, 5′-CAGGCUGUCAUGCAUUACATT-3′, 5′-GACGAAGGUUCAACAUCAATT-3′, 5′-GCGGCAGAAGAAAGACAAUTT-3′, and 5′-CCGAGACCUAUGGGAACAATT-3′ while a pool of 3 specific sequences were used for LC3B gene LC3B, 5′-GCCCUCUACUGAUUGUUAATT-3′, 5′-CUCCCUAAGAGGAUCUUUATT-3′, and 5′-GCCUGUGUUGUUACGGAAATT-3′.
A total of 105 cells were seeded in 6-well plates and transfected according to the manufacturer’s protocol for HiPerfect Transfection Reagent (Qiagen, USA). The transfection reagents were added to the cells (after diluting in 2 mL DMEM) for 24 h and then replaced by DMEM for another 24 h. Finally, RNA silencing was confirmed using Western blot and immunofluorescence analyses.
Cytotoxicity assays
The AlamarBlue® assay (Thermo Fisher Scientific, USA) was used for measuring cytotoxicity. This assay is a reliable method for determining cell viability9 and involves the reduction of resazurin (oxidized form, 7-hydroxy-3H-phenoxazin-3-1-10-oxide) to resorufin by cellular metabolic activity. Next, the number of cells with active mitochondria were counted, given that resazurin reduction is performed by mitochondrial enzymes12. This constitutes a simple, reliable, nontoxic, and safe method of measuring cell viability following irradiation13. After incubating for 7 h, fluorescence was measured using 590 nm as emission wavelength and 530–560 nm as excitation wavelength. Additionally, the culture medium was used as negative control while vitamin C was used as positive control for the full reduction of resazurin. Calculation of the relative fluorescence units and absolute % AlamarBlue reduction has been previously described13-15.
The advantage of the AlamarBlue method is that multiple measurements of the same well can be taken without killing the cells; thus, the viability time course of the same cell population can be monitored up to day 7 when nadir occurs or up to day 12 when there is a clear estimate of regrowth rates. As AlamarBlue is based on mitochondrial function, a disadvantage is that radiation or gene silencing may confound the method’s accuracy by interfering with mitochondrial function and should be excluded. Therefore, cell viability was assessed in parallel using an additional method employing the CyQUANT Cell Proliferation Assay Kit (C7026; Invitrogen), which is based on the fluorescence of a dye bound to cellular nucleic acids16. This assay provides a direct index of DNA content in viable cells attached to the bottom of the plate. However, the assay requires harvesting the cells before measurement, a disadvantage compared to the AlamarBlue assay. Neither radiation nor the siRNAs and short hairpin RNAs (shRNAs) employed had any impact on the accuracy of the AlamarBlue assay as it provided identical results to the CyQUANT method. Therefore, data from the AlamarBlue assay are reported herein.
Cell irradiation and dose/response curves
U87 and T98 cell lines were initially characterized regarding cell viability after exposure to single and multiple doses of irradiation (2–9 Gy). The radiation dose was given as a single fraction 24 h after transfection with siRNA. Both cell lines were evaluated for their radiosensitization potential after transfection with LC3A, LC3B, and TFEB siRNAs. Cells were seeded at a density of 250 cells/well in a 96-well plate. Cell irradiation was performed using a 6-MV beam produced by a linear accelerator (Precise Treatment SystemsTM, Elekta, Sweden) equipped with a multileaf collimator according to a previously described protocol regarding multidose irradiation of well columns within the same 96-well plate17. Cell viability was determined using the AlamarBlue assay. As cell count nadir occurs around the 7th day, cell counts at this time point were used to create dose/response curves9,17.
Chemosensitization
SiRNA technology was employed in combination with two anticancer drugs, temozolomide and cisplatin, to evaluate the effect when specific autophagy-lysosomal genes are silenced. LC3A, LC3B, and TFEB siRNAs were used to transfect U87 and T98 cells as described above. Cells (103 cells/well) were seeded in a 96-well plate and incubated with 100 μM temozolomide (Ridoca®; Enorasis AE, Greece) and 10 μM cisplatin (Platosin®; Pharmachemie Teva, Thailand) for 24 h. Cisplatin is not currently used for treatment of brain tumors but has been extensively used in combination with carmustine prior to temozolomide discovery. Drug efficacy was estimated in siRNA-transfected cells versus control cells and cell viability was determined using the AlamarBlue assay.
Time points of post-irradiation assays
The post-irradiation time point of 7 days was chosen for Western blot, microscopy, and quantitative PCR (qPCR) according to our extensive research on post-irradiation cell viability and regrowth, as this is the time point when the cell count nadir occurs and regrowth begins14. The day 2 time point was chosen to study an intermediate time point between irradiation and regrowth.
RNA isolation, cDNA synthesis, and qPCR
After irradiation with 4 Gy, total RNA of untreated and irradiated cells was isolated at 2 and 7 days using Nucleospin® RNA Plus Kit (Macherey-Nagel, Germany). RNA quality (260/280 nm and 260/230 nm ratios) as well as quantity was evaluated and performed using the NanoDrop 2000c (Thermo Fisher Scientific).
Total RNA (500 ng) was used to synthesize cDNA using the PrimeScriptTM RT Reagent Kit (Takara, Japan) according to manufacturer’s instructions. The reaction steps included incubation at 37 °C for 15 min and 85 °C for 5 s. Both steps were performed in a thermal block cycler with a heated lid (Mastercycler® pro, Eppendorf, Germany).
qPCR was performed in triplicate using the KAPA SYBR® FAST qPCR Kit optimized for LightCycler® 480 (KR0392—v7.14; Kapa Biosystems) on a LightCycler® 480 Instrument II (Roche). qPCR primer sets were designed using the Roche primer design tool (https://lifescience.roche.com/en_gr/brands/universal-probe-library.html#assay-design-center) as follows, human MAP1LC3A forward, 5′-CATGAGCGAGTTGTTCAAGA-3′ and reverse, 5′-CCATGCTGTGCTGGTTCA-3′; human MAP1LC3B forward, 5′-CGCACCTTCGAACAAAGAG-3′ and reverse, 5′-CTCACCCTTGTATCGTTCTATTATCA-3′; human LAMP2a forward, 5′-GCTCGTTCTGGTCTGCCTA-3′ and reverse, 5′-TGTCAAATTAAGTTCCAATGCATAA-3′; human SQSTM1/p62 forward, 5′-AGCTGCCTTGTACCCACATC-3′ and reverse, 5′-CAGAGAAGCCCATGGACAG-3′; human TFEB forward, 5′-AACATGACAGCAAGCTCAGG-3′ and reverse, 5′-CTGCATGCGCAACCCTAT-3′; human cathepsin D (CTSD) forward, 5′-CATCTTCTCCTTCTACCTGAGCA-3′ and reverse, 5′-GTCTGTGCCACCCAGCAT-3′; human HPRT1 forward, 5′-TGACCTTGATTTATTTTGCATACC-3′ and reverse, 5′-CGAGCAAGACGTTCAGTCCT-3′. All sets of primers were designed and purchased from Roche.
Cycling conditions for qPCR consisted of four steps; pre-incubation at 95 °C for 3 min, amplification for 40 cycles at 95 °C for 10 s, 56 °C for 20 s, and 72 °C for 1 s, melting curve steps at 95 °C for 5 s and 65 °C for 1 min, and cooling at 40 °C for 10 s. Quantitative analysis was performed using the LightCycler software (LightCycler® 480 SW 1.5.0 SP4 v1.5.0.39, Roche, Switzerland), where cycle thresholds (Cts) were determined for the genes of interest and then normalized to HPRT1 (measured at specific time points of 0 h, 2 days, and 7 days) using the ΔΔCt method. The experiments were performed in triplicate and repeated three times.
Western blot analysis
Glioma cell lines (U87MG and T98G) from ATCC were cultured under standard conditions in a 5% CO2 incubator at 37 °C. Cells lysates were collected 48 h and 7 days after irradiation using a sucrose buffer (0.25 M sucrose and 25 mM Tris-HCl) and then homogenized using a plastic pestle. Next, the samples were centrifuged at 15,000 × g for 15 min to separate membrane, nuclear, and cytoplasmic proteins. Total protein concentration was calculated using the BCA Protein Assay Kit (Thermo Fisher Scientific).
Of each protein, 40 μg were loaded for analysis. Protein samples were separated on discontinuous SDS gels using an 8% separating gel for LAMP-2a, 10% for SQSTM1/p62, TFEB, CTSD, and caspase-9, and 12.5% for LC3A and LC3B. Additionally, 5% stacking gels were used and 40 ng of sample was analyzed on the gel. Electroblotting was performed with Porablot PVDF membranes (Macherey Nagel, Germany) and then membranes were blocked with 5% non-fat dry milk in 150 mM NaCl, pH 7.5 10 mM Tris (TBS), and 0.1% (v/v) Tween 20 at 20–25°C for 2 h followed by overnight hybridization with primary antibodies at 4 °C. The membranes were then hybridized for 2 h at room temperature with the secondary antibodies goat anti-rabbit IgG-HRP (1:3,000; #1706515; Bio-Rad Laboratories, USA) or goat anti-mouse IgG-HRP (1:3,000; #1706516; Bio-Rad Laboratories). Finally, membranes were developed using the ChemiDoc MP Imaging System (Bio-Rad Laboratories, CA, USA).
Primary antibodies used were rabbit polyclonal anti-LC3A (1:15,000; ab62720; Abcam, UK), mouse monoclonal anti-LC3B (1:1,000; 0231-100/LC3-5F10; NanoTools, Germany), rabbit polyclonal anti-SQSTM1/p62 (1:1,000; ab64134; Abcam, UK), rabbit polyclonal anti-TFEB (1:2,000; A303-673A; Bethyl Laboratories, Inc., TX, USA), mouse monoclonal anti-CTSD (CTD-19; 1:1,000; ab6313; Abcam, UK), rabbit polyclonal anti-LAMP2a (1:1,000; ab18528; Abcam, UK), and rabbit polyclonal anti-caspase 9 (1:2,000; ab47537; Abcam, UK).
Each blot was then stripped, dried overnight, re-hybridized with rabbit polyclonal anti-beta actin (1:5,000; ab75186; Abcam, UK).
All experiments were performed three times. Blot images were captured using the ChemiDoc MP Imaging System and densitometric analysis of proteins was performed using the Image Lab software (Bio-Rad Laboratories, CA, USA).
Confocal immunofluorescence
For immunofluorescence staining, cells were grown on No. 1.5 glass coverslips (Sigma-Aldrich, MO, USA), fixed in 3.7% formaldehyde/phosphate buffered saline (PBS) (pH 7.4) for 20 min at 37 °C, and then permeabilized in PBS/0.1% v/v Triton X-100 (pH 7.4) for 5 min at room temperature. Cells were blocked in PBS/5% w/v bovine serum albumin (BSA) (pH 7.4) and then stained with various combinations of the following antibodies for 1 h at room temperature, rabbit polyclonal anti-MAP1LC3A (1:500; ab62720; Abcam, UK), mouse monoclonal anti-LC3B (1:100; 0231-100/LC3-5F10; NanoTools, Germany), mouse monoclonal anti-p62 (1:100; ab56416; Abcam, UK), rabbit polyclonal anti-LAMP2A (1:500; ab18528; Abcam), mouse monoclonal anti-CTSD (1:200; ab6313; Abcam, UK), rabbit polyclonal anti-caspase-9 (1:200; ab47537; Abcam, UK), rabbit polyclonal anti-TFEB (1:500; A303-673A; Bethyl Laboratories, TX, USA), and rabbit polyclonal anti-gamma H2A.X (1:500; ab11174; Abcam, UK). Cells were washed in pH 7.4 PBS, incubated with the appropriate CF488A or CF568 secondary antibodies (1:500; Biotium) at room temperature, and then DNA was counterstained with Hoechst 33342 (1 μg/mL; Sigma-Aldrich,). After final washes, the coverslips were mounted in an in-house Mowiol mounting medium. Imaging was performed using a customized Andor Revolution Spinning Disk Confocal System built around a stand (IX81; Olympus) with a 100 ×, 1.4 numerical aperture lens and a digital camera (Andor iXon+885) at the Cell Imaging and Biomolecular Interaction (CIBIT) facility at Molecular Biology and Genetics-Democritus University of Thrace, Greece (MBG-DUTH). Image acquisition was performed using Andor IQ 2 software. Optical sections were recorded every 0.3 μm. All confocal microscopy images presented in this work are two-dimensional (2D) maximum intensity projections of z-stack images acquired using ImageJ software (v1.47; National Institute of Health, USA). Image intensity analysis of the obtained data sets was performed using ImageJ software and custom image processing macros were developed to quantify protein levels (fold increase of fluorescence intensity compared to control). Graphs were plotted using the GraphPad Prism statistical package (v5.01; GraphPad Software Inc., USA).
Aggresome detection using confocal microscopy
Aggresome visualization was performed using the PROTEOSTAT® Aggresome detection kit (Enzo) according to manufacturer's instructions. Aggresome accumulation can be directly linked to inhibition of autophagy, specifically inhibition of autophagic flux. MG132, a proteasome inhibitor that promotes aggresome accumulation, was used as a positive control and nuclei were counterstained using Hoechst 33342—both chemicals were provided by the kit. Samples stained with the PROTEOSTAT aggresome dye were imaged using confocal microscopy at 60x magnification using a standard red filter.
Apoptosis detection using fluorescence microscopy
Apoptosis was detected with annexin V (green fluorescence Excitation/Emission, Ex/Em = 490/525 nm)—an indicator that binds cell surface phosphatidylserine—using the Apoptosis/Necrosis Detection Kit (blue, green, red; ab176749; Abcam) and Cell-IQ MLF live imaging system. The detection kit was used according to manufacturer’s instructions while omitting the blue dye for live cells due to the lack of an appropriate excitation light source. Untreated cells were used as negative and cells treated for 24 h with 1 μM docetaxel, an anticancer taxane drug, were used as positive control for apoptosis induction. T98G and U87MG cancer cell lines were irradiated with 4 Gy and apoptosis was monitored over several time points at 2 h, 4 h, and 24 h and 48 h. Fluorescent images were processed and merged after which fluorescence intensity was quantified using ImageJ and graphs were plotted using GraphPad Prism. Additionally, percentage of cells with apoptotic features was subjectively assessed and plotted.
shLC3A stable transfected cell lines
Specific shRNA sequences for targeting LC3A were designed by and purchased from GenePharma (Shanghai, China). T98G and U87MG cell lines were stably transfected with shLC3A plasmids carrying the geneticin resistance gene with a pool of 4 shRNA sequences as follows, 5′-CACCGCGAGTTGGTCAAGATCATCCTTCAAGAGAGGATGATCTTGACCAACTCGCTTTTTTG-3′, 5′-CACCGCTGTAAGGAGGTACAGCAGATTCAAGAGATCTGCTGTACCTCCTTACAGCTTTTTTG-3′, 5′-CACCGGTGATCATCGAGCGCTACAATTCAAGAGATTGTAGCGCTCGATGATCACCTTTTTTG -3′, and 5′-CACCGCTTCCTCTATATGGTCTACGTTCAAGAGACGTAGACCATATAGAGGAAGCTTTTTTG-3′.
The cells were plated in 6-well plates and incubated for 24 h and transfected with 4 μg of the relevant shRNA plasmids in DMEM medium (without any antibiotics and FBS) using Metafectene Pro (Biontex Laboratories GmbH, Germany) according to manufacturer’s instructions. The medium was then replaced with fresh DMEM 6 h after transfection to prevent extensive cytotoxicity and cell stress. The medium was replaced with DMEM containing 0.5 mg/μL geneticin (Abcam) 24 h after transfection, after which the cells were cultured in this medium for 15 days until the selection process for cells carrying the shLC3A plasmids (providing geneticin resistance) was complete; this was confirmed by the death of control/untransfected cells that were not resistant to geneticin.
Senescence-associated-β-galactosidase staining (SA-β-gal)
T98shLC3A and U87shLC3A glioblastoma cells and their paternal T98 and U87 cells, respectively, were irradiated with 4 Gy using a Cobalt-60 gamma-ray irradiator (TheratronTM, Theratronix, Canada). Untreated cells were used as negative control. Staining for SA-β-gal activity was performed as previously described18 with minor modifications. Briefly, 2–3 × 104 cells were seeded on 6-well plates, irradiated with 4 Gy after 24 h, and fixed after 4 days. Cells were then washed twice with PBS, fixed in 0.2% glutaraldehyde/2% formaldehyde for 5 min, washed again with PBS, and incubated in the absence of CO2 at 37 °C for 16 h with freshly made staining solution [150 mM NaCl, 2 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 20 mM citric acid, and 40 mM Na2HPO4, pH 6.0] containing 1 mg/mL of 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal). The staining solution was removed the following day and then cells were washed twice with PBS. The total number of cells and cells positive for SA-β-gal staining were counted in 12 randomly chosen fields per condition.
Animal care and xenograft experiments
Animal care and handling was carried out according to the guidelines set under Directive 86/609/EEC. All procedures were approved by the Veterinary Direction for Animal Research of the Department of Experimental Surgery at the Democritus University of Thrace, Greece. Athymic nude mice were housed at the Easy Flow IVC Air Handling Unit (Tecniplast), which is comprised of individually ventilated cages. Caging included sterile bedding conditions as well as diet and water provided ad libitum. The animals were maintained at an ambient temperature of 23 °C and under 12 h light:12 h dark photoperiods.
The study was approved by the Committee of Evaluation of Experimental Animal Research Protocols and by the local Veterinary Authorities. Athymic nude mice (Hsd:Athymic Nude-Foxn1nu) were purchased from Harlan® Laboratories. Mice (8-week-old) were randomly assigned into two groups, one for each cell line. Mice in both groups were injected subcutaneously with 107 U87 or T98 cells; wild type and shLC3A-transfected cells were injected dorsally into the left and right hindlimbs, respectively. The timing of injections was chosen based on previous experiments that established growth kinetics of the cell lines in order to perform abdominal irradiation when both tumors’ volumes (wild type and shRNA) are approximately 1 cm3 in diameter.
Cell injection preparation involved centrifuging cells, recording dispersed cell pellet volume (< 200 μL), and then adding an equal volume of Cultrex Pathclear® Basement Membrane Extract (BME) Type 3 (3632-005-02; Trevigen®, USA) to facilitate cell adherence to the subcutaneous injection space and to consolidate tumor formation.
Animals were monitored daily after implantation of CCL-xenografts and received partial body (lower body) irradiation with 6 Gy/day (using a 6-MV LINAC) for two consecutive days until tumors reached approximately 1 cm × 1 cm in size. Irradiation was directed to the lower mouse body where the tumors were growing using a customized apparatus and 3 dimensional (3D) radiotherapy planning after computed tomography (CT)-scanning of mice19,20. Tumor growth was monitored for 30 consecutive days post-irradiation.
Statistical analysis
Statistical analysis and graphical representation were performed using GraphPad Prism. Paired and unpaired two-tailed t-tests were used for comparisons where appropriate. A P < 0.05 indicated significant difference.
Results
Effect of radiation on autophagic flux
Protein levels of LC3A-II and LC3B-II in the T98 cell line, assessed by Western blot using the pellet fraction, showed a clear reduction 2 days post-irradiation with 4 Gy that returned to normal levels by day 7 (Figure 1A, B, and Figure S1). This finding may indicate the consumption of autophagosomes as a result of increased autophagic flux and is supported by a concurrent reduction in p62 expression levels (Figure 1A, B). Aggresomes were not found accumulated, which is compatible with functional autophagic flux (Figure 1C).
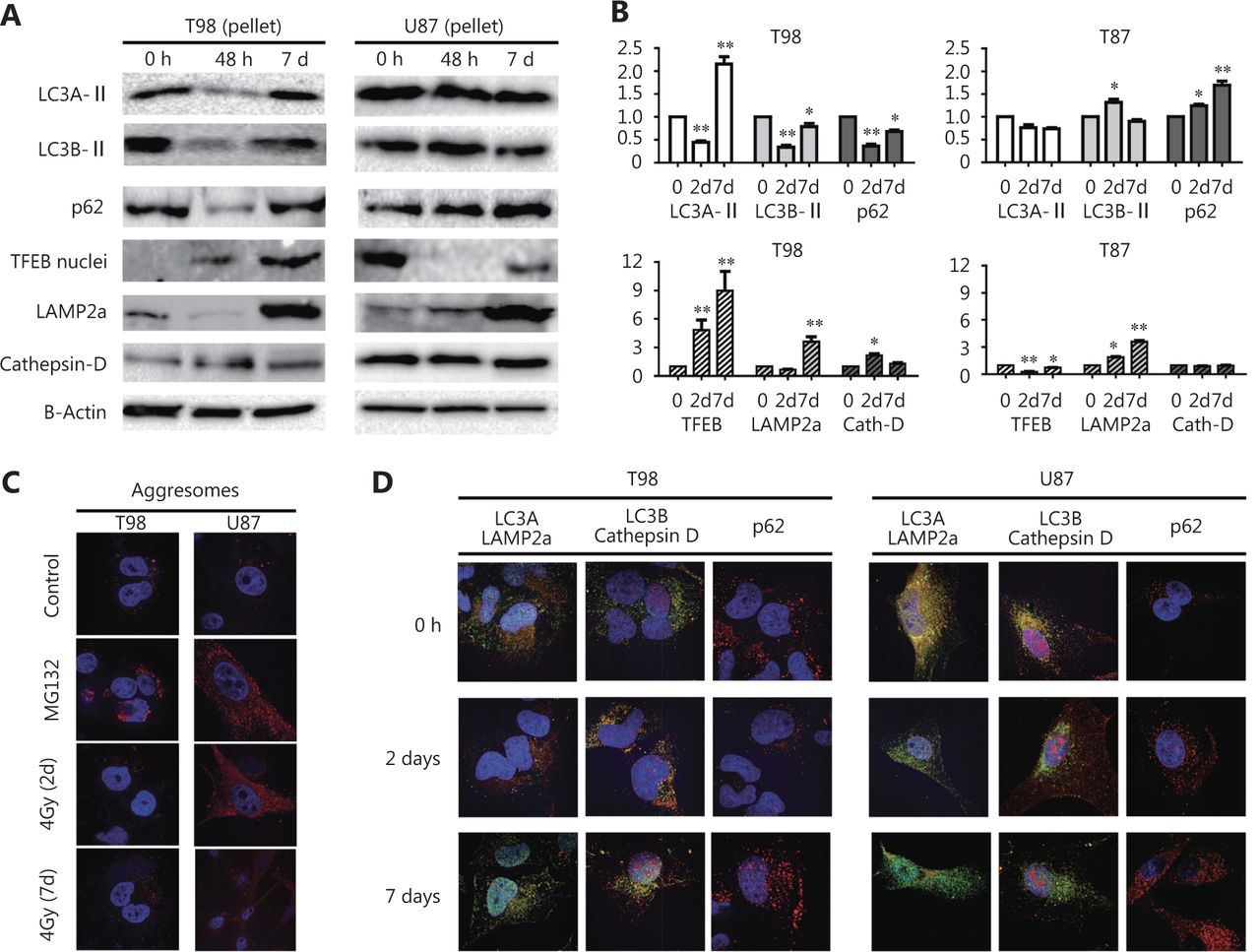
(A) Western blot images of the response of T98 and U87 cell lines, 2 and 7 days after 4Gy irradiation, examined in the pellet fraction (where the autophagosomes reside) for LC3A (16 KDa), LC3B (16 KDa), p62 (60 KDa), LAMP2a (100 KDa) and CTSD (34 KDa), and in the nuclear fraction for TFEB (55 KDa). (B) Band densitometry of the Western blots (performed in triplicates). The Y-axis shows the fold increase compared to time point 0 h (*P < 0.05, ** P < 0.01, *** P < 0.001). (C) Typical images (100 x) of aggresome (red) accumulation following exposure of cells to the proteasome inhibitor MG132 (positive control) and 4 Gy irradiation. (D) Confocal microscopy images (100 x) of LC3A (green)/LAMP2a (red), LC3B (red)/CTSD (green) colocalization and of p62 (red) protein expression, following 4 Gy irradiation.

Western blot images of the LC3A and LC3B proteins, in the pellet fraction of the T98 and U87 glioblastoma cells, 48 h and 7days after exposure to 4Gy of radiation.
On the other hand, a different pattern of autophagic response was observed for the U87 cell line. Western blot analysis of cell pellets following 4 Gy irradiation showed stable LC3A-II and increasing LC3B-II levels by day 2 in parallel with increasing p62 levels up to 7 days after irradiation, suggesting a protracted blockage of autophagic flux (Figure 1A, B). Aggresome accumulation after irradiation was evident in the U87 cell line and persisted up to 7 days (Figure 1C).
This divergent response pattern between the two cell lines was confirmed by confocal microscopy where autolysosomal fusion—detected via LC3A/LAMP2a and LC3B/CTSD colocalized expression—was sustained after irradiation of the T98 cell line but was sharply reduced in U87 cells (Figure 1D). Of note, the pre-irradiation colocalization levels were far more intense in U87 than in T98 cells, suggesting that the blocking effect of radiation is not dependent on the basal levels of autophagic flux. p62 protein expression was found reduced 2 days after irradiation of T98 cells, whereas expression increased in U87 cells, further supporting the divergent susceptibility of autophagic machinery to radiation between the two cell lines (Figure 1D). The LC3A/LAMP2a and LC3B/CTSD colocalization pattern in T98 and U87 cells in response to bafilomycin, a known agent that blocks autophagy flux, is shown in Figure S2.

In order to confirm that LC3A-LAMP2Aor LC3B-CTSD imaging indeed records “autophagic flux” changes, T98 and U87 cells were incubated with Bafilomycin A, (100 nM) for 24h. The autophagosome and lysosome fusion was disrupted as it is demonstrated in the relevant confocal microscopy images. As the drug has its major effect on slowing the degradation of LC3-II within existing autolysosomes, incubation for 24h results to accumulation of LC3A or LC3B auto-lysosomes. This, however, was more prominent in the T98 cell line.
Effect of radiation on lysosomal protein kinetics
Regarding lysosomal markers, expression levels of LAMP2a in the cell pellet fraction declined 2 days after irradiation of the T98 cell line, indicating increased lysosomal consumption (Figure 1A, B). In contrast, LAMP2a expression levels gradually increased for 7 days after irradiation of U87 cells, supporting the postulated blockage of autophagic flux.
The nuclear localization of TFEB, which is involved in lysosomal biogenesis, was found increased at day 2 and 7 in the T98 cell line (Figure 1A, B), indicating a cellular reaction of adapting to the increased lysosomal demands induced post-irradiation. On the other hand, nuclear levels of TFEB dropped in U87 cells (Figure 1A, B). CTSD levels, however, slightly increased after irradiation in the T98 cell line only (Figure 1A, B), which is in contrast to the kinetics of LAMP2a as a marker of lysosome density. Whether this reflects the appearance of lysosomes with increased CTSD content stimulated by early nuclear overexpression of TFEB in response to radiation is a hypothesis that demands further investigation.
qPCR analysis
qPCR showed minor changes in autophagosomal and lysosomal mRNA levels 2 days after irradiation of T98 and U87 cells. By day 7, an induction in gene expression levels of LC3A, p62, and the three lysosomal genes studied was observed, which was stronger in T98 than in the U87 cell line, suggesting transcriptional changes that support the autophagic machinery (Figure 2A).
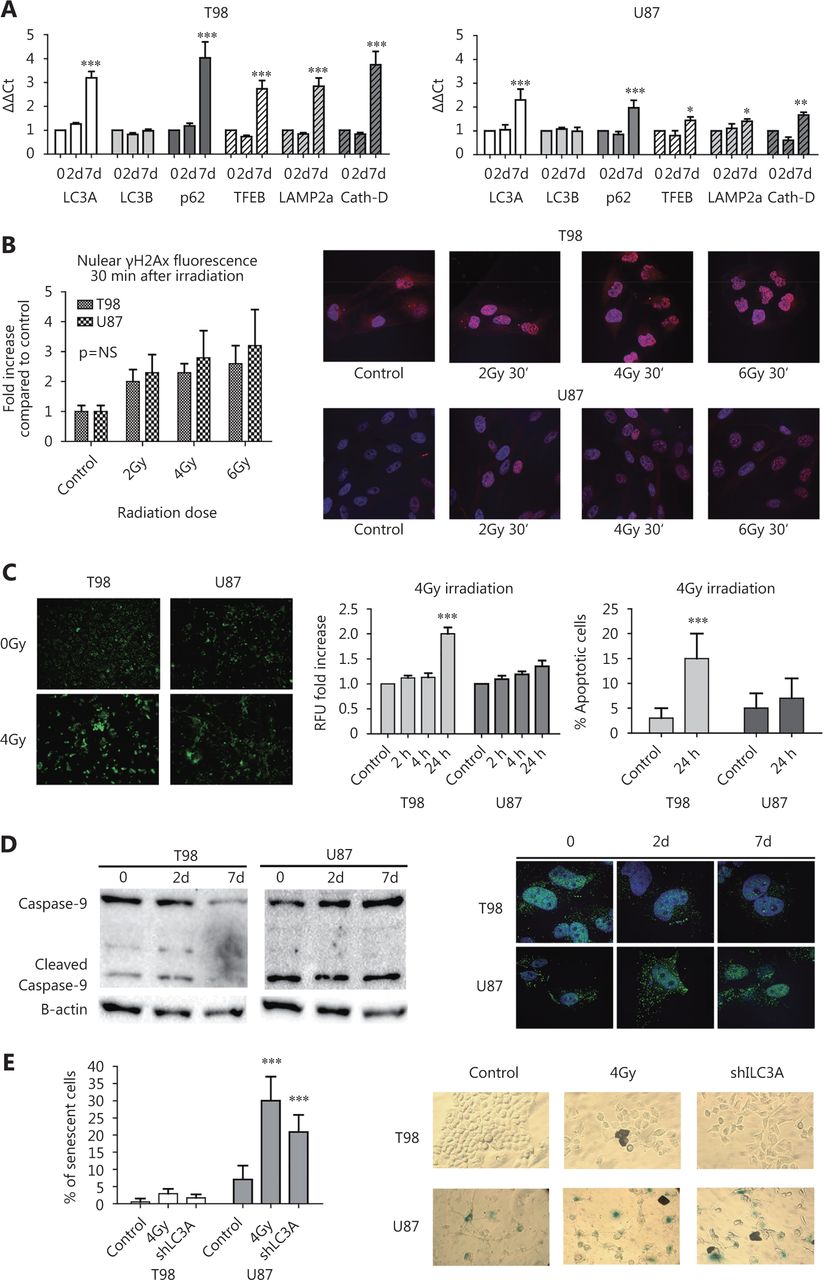
(A) Relative quantitative analysis of mRNA levels of the LC3A, LC3B, p62, TFEB, LAMP2a and CTSD mRNA levels at 0, 2 and 7 days after exposure of T98 and U87 cells lines to 4 Gy of radiation (*P < 0.05, **P < 0.01, ***P < 0.001). (B) γH2AX nuclear foci expression and image densitometry of nuclear staining of T98 and U87 cell lines exposed to escalated doses of radiation. (C) Detection of apoptosis in T98 and U87 cell lines, assessed with confocal microscopy, following 4 Gy of irradiation, at various time points (typical confocal images, fluorescence intensity assessment and % of apoptotic cells) (40 x). (D) Caspase 9 (csp9) expression levels in the T98 and U87 cell lines, at 2 and 7 days, following exposure to 4 Gy radiation. (E) Percent of cells with SA-β-gal expression in wild type T98 and U87 cells (control), cells with silenced LC3A (shLC3A cells), and following exposure to 4 Gy of radiation (on the 4th day).
Effect of radiation on DNA damage
γH2AX-positive nuclear foci increased 30 min after irradiation and their accumulation was proportional to the radiation dose. Baseline levels of γH2AX foci were higher in T98 cells; however, the fold increase following irradiation was slightly higher in U87 than in T98 cells, but the difference was not statistically significant (Figure 2B).
Effect of radiation on apoptosis
At 24 h and 48 h after irradiation (but not earlier) with 4 Gy, T98 cells exhibited a significantly higher number of apoptotic cells compared with control cells. In contrast, the number of apoptotic cells did not increase in U87 cells (Figure 2C).
Effect of radiation and LC3A silencing on senescence
In T98 cells, caspase-9 levels were found reduced after 4 Gy irradiation (2.5- and 10-fold decrease by day 2 and 7, respectively), while the opposite pattern was exhibited in U87 cells (3.5- and 8-fold increase by day 2 and 7, respectively; Figure 2D). SA-β-gal staining indicated that there was a minor effect on senescence induction in T98 cells following irradiation with 4 Gy or LC3A gene silencing where approximately 3% and 2% of cells were senescent, respectively. In contrast, a strong induction of senescence (approximately 30% and 20% of cells, respectively) was observed in U87 cells (Figure 2E).
Autophagy blockage and radiosensitization in vitro
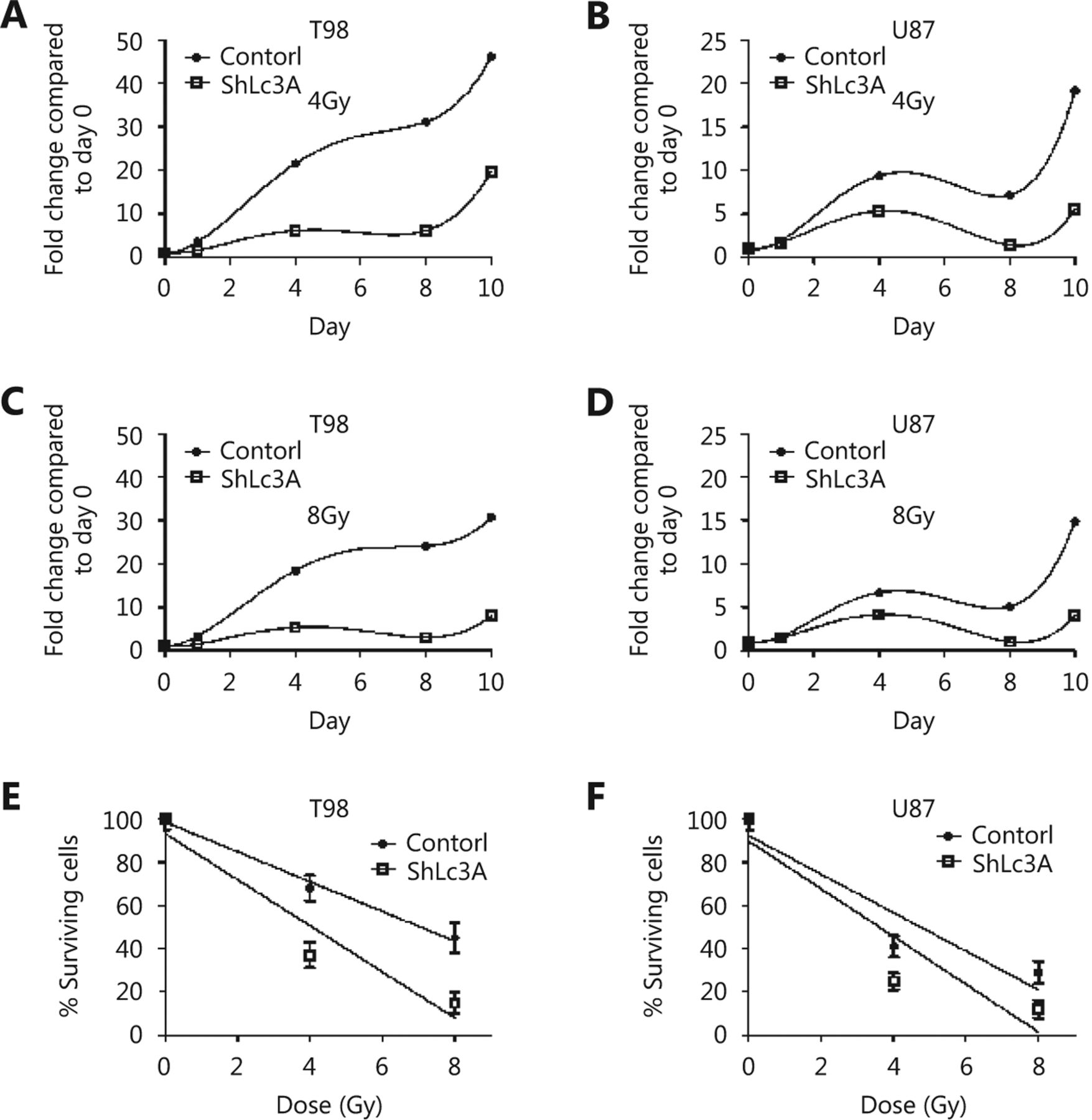
Using specific siRNAs for targeting LC3A, LC3B, and TFEB, we investigated the role of these genes in radiosensitivity of the two glioblastoma cell lines. Confocal microscopy and Western blot analysis of the supernatant fraction showed strong gene silencing in whole cell fractions (Figure 3A). LC3A, LC3B, and TFEB protein levels showed a 15-, 12-, and 8-fold decrease in T98 cells, respectively, and 14-, 11-, and 6-fold decrease in U87 cells, respectively.

(A) Confocal images (100 x) and Western blot analysis of T98 and U87 glioblastoma cell lines stably transfected with shLC3A, shLC3B and shTFEB, showing successful suppression of protein expression (assessed in the supernatant fraction). (B) Growth delay dose/response curves, assessed on the 7 th day after irradiation, in the T98 and U87 cell lines without and with repression of the LC3A, LC3B, LC3A/B and TFEB protein expression, using siRNA. (C) Cell viability of the T98 and U87 cell lines after 24 h exposure to temozolomide (10 μΜ) or cisplatin (10 μΜ), in wild type and transfected cells with siLC3A, siLC3B or siTFEB (*P < 0.05, ** P < 0.01, *** P < 0.001).
Following single fraction exposure to escalated doses of irradiation, we calculated RD50, the radiation dose needed for 50% growth suppression on day 7 (when cell count nadir occurs). RD50 was 4.2 Gy and 5.9 Gy for U87 and T98 cells, respectively, indicating that T98 cells are relatively more radioresistant than U87 (Figure 3B). Notably, suppression of cell viability at 2 Gy (standard fractionation used in the clinical practice) was 13% in T98 and 27% in U87 cells, demonstrating a shallow shoulder of the survival curve for T98 cells, which supports the increased radioresistance of T98 cells to low dose per fraction radiotherapy.
Both cell lines were sensitized after silencing of LC3A, LC3B andTFEB genes (P<0.01,Figure 3B). Silencing of TFEB gene expression exhibited the strongest radiosensitizing effect, where RD50 dropped to 1.2 Gy and 1.9 Gy for T98 and U87 cells, respectively (P < 0.001). Combined silencing of LC3A and LC3B gene expression was necessary to produce a similar radiosensitizing effect on T98 cells as TFEB silencing. For U87 cells, double silencing of LC3A and LC3B did not enhance sensitization compared with single silencing. Overall, silencing of autophagy genes produced a more potent sensitizing effect on the radioresistant T98 cell line than on U87 cells ( Figure 3B).
Autophagy blockage and chemosensitization
A concentration of 100 μM and 2 μM was chosen for temozolomide and cisplatin, respectively, following a dose escalation experiment to define the appropriate doses for achieving approximately 80% control cell viability after 24 h drug exposure. Silencing of LC3A, LC3B, or TFEB resulted in significant sensitization to temozolomide but not to cisplatin (Figure 3C).
Autophagy blockage and radiosensitization in vivo
U87shLC3A and T98shLC3A stable transfected cell lines were established and Western blot analysis of the supernatant fraction showed strong reduction of the LC3A-I protein (Figure 4A). Two groups of five athymic mice for each cell line were used in this experiment. Following cell implantation, U87shLC3A and T98shLC3A cells exhibited a slower growth rate compared with wild type U87 and T98 cells, respectively (Figure 4B, C); this effect was more pronounced with the T98 cell line. The reduced growth rates of wild type and tranfected cells was also confirmed by in vitro experiments (Figure 3S). Moreover, prior to in vivo experiments, the radiosensitivity of shLC3A cells was tested in vitro compared with untransfected cells (Figure S4).

(A) Reduced expression of the LC3A-1 protein in the supernatant of T98 and U87 cell lines, following transfection with specific vs. random non-coding siRNAs. (B, C) Growth kinetics of the wild type and shLC3A transfected T98 (B) and U87 cell lines (C) following subcutaneous implantation in athymic mice. (D) A typical image of a U87 implanted tumor in an athymic mice showing different size of the wild type and shLC3A implanted tumors, 12 days after irradiation.
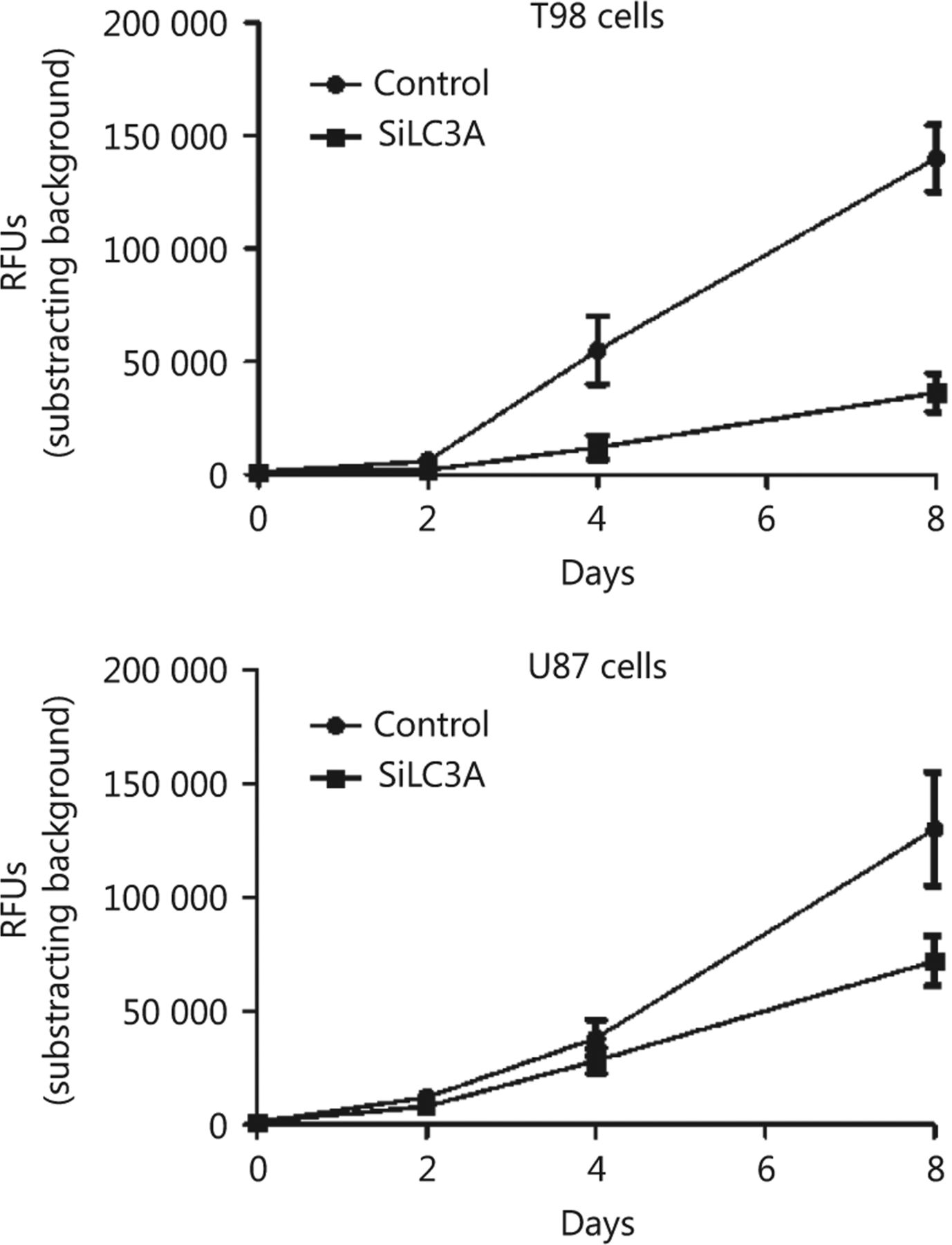
In vitro growth curves of T98 and U87 control cells and cells transfected with LC3A shRNA.

Growth curves of T98 glioblastoma cell lines following exposure to 4 Gy (A) and 8 Gy (B) and dose response curves (C). Growth curves of U87 glioblastoma cell lines following exposure to 4 Gy (d) and 8 Gy (e) and dose response curves (f).
When tumors reached a median of 1 cm2 in area, they were irradiated with two consecutive daily fractions of 6 Gy. Wild type, U87 and T98, showed growth at a reduced rate and then entered an accelerated phase of growth by day 12 and 20, respectively. The size of T98shLC3A tumors showed a dramatic reduction and were undetectable at 18 days irradiation (Figure 4B). U87shLC3A tumor size remained stable after irradiation and throughout the 30 days of observation (Figure 4C). The difference isn growth rate reduction between wild type and transfected cells was statistically significant at all post-irradiation time points (P<0.001). A typical macroscopic image of a U87 xenograft at 12 days after irradiation is presented inFigure 4D, showing the difference between wild type and shLC3A tumors.
Discussion
Glioblastoma is a very aggressive type of cancer that remains one of the most lethal human tumors, despite its localized and non-metastasizing growth pattern and advances in radiotherapy13. High doses of radiotherapy, at the limits of normal brain tolerance, fail to eradicate the cancer and thus 95% of tumors relapse within 1–3 years post-treatment. Temozolomide marginally improves survival figures by some months3, while angiogenesis or epidermal growth factor receptor (EGFR) inhibitors have failed to show any clinical benefits12. Experimental studies suggest that the remarkable radioresistance of glioblastoma cells is due to their reluctance in triggering apoptotic pathways. Loss of PTEN or of other apoptosis-related genes may be involved in this phenomenon9. Moreover, several studies have suggested that autophagic cell death is the preferred death pathway for glioblastoma cells19,20,21. Interfering with this biological pathway may prove to be the key for developing more effective radiosensitizing strategies8.
Therefore, the first step is studying the basal levels of autophagic flux and its response to radiation. In our previous immunohistochemical study, we found that autophagy markers are overexpressed in approximately half human glioblastomas and provided evidence that overactive autophagy counteracts the response of these tumors to radiation23. Therefore, we examined autophagic flux kinetics after 4 Gy X-ray irradiation of two common glioblastoma cell lines, T98 and U87, in this study. T98 cells sustain PTEN expression, while U87 cells exhibit complete loss of expression of the protein, as we previously reported23. PTEN is an anti-oncogene that negatively regulates the Akt-pathway and PTEN loss is expected to be linked with reduced apoptotic tendency and resistance to radiotherapy24. Indeed, this was confirmed in the current study as the percentage of apoptotic cells did not increase following irradiation of the U87 cell line, whereas a clear increase was observed in T98 cells. However, despite the decreased apoptotic response of U87 cells, they were more radiosensitive compared with T98 cells. In addition, there was no significant difference in DNA repair abilities between the two cell lines, as evidenced by similar intensity of γH2AX nuclear foci formation at 30 min after irradiation. These findings strongly support the hypothesis that the radiosensitivity of glioblastoma cell lines is apoptosis-independent.
To investigate the involvement of autophagic response in the radioresistance levels of these two glioblastoma cell lines, the cells were exposed to 4 Gy of radiation. A differential response in terms of autophagy and lysosomal protein expression kinetics was observed between the two cell lines. The relatively radioresistant T98 cell line showed an intensification of autophagic flux of 2 days after irradiation, while the U87 cell line exhibited a blockage of autophagic flux that extended up to 7 days post-irradiation. It should be noted that the U87 cell line had an increased baseline autophagic flux, as evidenced by the extensive colocalization of LC3s with lysosomal markers observed by confocal microscopy. This is in agreement with our previous study where U87 cells expressed remarkably high levels of autophagy and lysosomal proteins compared with T98 cells23. Thus, it appears that radiation resistance is not dependent on baseline autophagic flux levels but rather on the ability of cells to sustain or intensify autophagy following irradiation. This finding has also been reported in the radioresistant prostate cancer cell line PC325. Notably, an increase in transcription of autophagy and lysosomal genes characterized the radioresistant T98 cell line, with TFEB nuclear increased shift dominating the response as early as 2 days post-irradiation.
The above findings may explain the discrepancy found among translational studies. Some studies have suggested that reduced expression of L4C3B or of Beclin-1—and thus glioblastomas with reduced autophagic activity—is linked with a more aggressive clinical course of the cancer26-28. Other studies have shown the exact opposite effect by linking high LC3B or LC3A expression with poor survival of glioblastomas23,29. Taking into account the findings of the current study, where radiotherapy outcomes depend on the specific autophagic flux response of tumors to certain radiation doses and not on the initial status of autophagic flux, it appears that quantification of baseline expression levels of autophagy proteins may not be a reliable predictive marker.
Whether glioblastoma cells intensify their autophagic response to radiation in a radiation dose-dependent manner in each individual cell system demands further investigation, as this may have important implications in the clinical fractionation of radiotherapy8. This may also explain the discrepancies reported in the literature, as the same radiation dose may enhance or repress autophagic flux in different cell types. Higher doses may repress autophagy in cells where lower doses allow intensification of the process11,22. Interestingly, a study by Palumbo et al.30 showed that low and intermediate doses of radiation induced higher Beclin-1 and Atg5 expression levels in T98 cells compared with high radiation doses, supporting the dose-dependent autophagic response to radiation.
The importance of sustained autophagic flux following exposure to radiation for cell survival was further supported by experiments that silenced key autophagy genes (LC3A and LC3B) and TFEB, which regulates lysosomal biogenesis7. Both T98 and U87 cell lines were strongly sensitized to radiation and silencing TFEB gene expression exhibited the strongest radiosensitizing effect. This is in agreement with previously published studies suggesting that blockage of autophagy sensitized glioblastoma cells to radiation31,32. A study by Lomonaco et al.33 provided evidence that inhibition of autophagy enhanced CD133+ glioblastoma stem cell sensitivity to ionizing radiation after bafilomycin treatment or Atg5 silencing. Notably, Palumbo et al. 30 found that Atg7 silencing resulted in reduced T98 cell sensitivity to radiation and to a combination of radiation with anti-EGFR therapy34, indicating a complex role of different Atgs in radiation response.
Sensitization was most potent in the T98 radioresistant cell line. Despite the fact that T98 cells had a lower baseline level of autophagic flux compared with U87 cells, they were able to intensify autophagic flux following exposure to 4 Gy radiation. Therefore, it seems that the strongest radiosensitizing effect of autophagy inhibition is related to blockage of the reparative autophagy mechanism triggered by radiation damage and not to baseline autophagy flux levels. U87 cells were also sensitized by autophagy silencing but sensitization efficacy was lower than in T98 cells, as radiation had already blocked autophagic flux. This was also verified by in vivo experiments where elimination of xenografts following irradiation of autophagy-impaired shLC3A transfected cells was only observed in T98 cells, whereas U87 cells exhibited a strong arrest in tumor growth rather than regression.
Whether autophagy blockage leads to senescence in PTEN-deficient cells is a hypothesis that emerged from the current data. Previous studies have shown that PTEN loss may result in increased murine double minute 2 (MDM2) degradation and p53 stabilization35,36, which may be responsible for a senescence response that resulted in the growth arrest observed in U87 xenografts. Induction of caspase-9 in U87 cells following irradiation, which was observed herein, may be an indicator of activation of senescence rather than apoptosis, as studies support the involvement of this pro-apoptotic protein in senescence induction37. Moreover, Sohn et al.38 found that irradiation-induced caspase-9 leads to senescence, and not apoptosis, in p21-proficient colon cancer cell lines. Senescence (Sen)-β-gal experiments confirmed that a large percentage of U87 cells entered senescence after blocking autophagy following exposure of cells to 4 Gy irradiation or by LC3A gene silencing. T98 cells were refractory to senescence induction and preferred to intensify death pathways, such as apoptosis. This finding may also explain the in vivo observation that U87shLC3A tumors entered a prolonged phase of growth arrest after irradiation, whereas T98shLC3A tumors were nearly eliminated after irradiation.
Another interesting finding was that silencing LC3 or TFEB genes sensitized both cancer cell lines to temozolomide, a key drug used in combination with radiotherapy for glioblastoma treatment. Katayama et al.39 previously showed that temozolomide induces autophagy in glioma cell lines in parallel with increased ATP production. This production of ATP was found to result from increased autophagy as production was suppressed by autophagy inhibitors; thus, enhancement of autophagic flux was suggested as a pathway linked with glioma resistance to temozolomide. In fact, combining bafilomycin, which blocks autophagy by preventing the fusion of mature autophagosomes with lysosomes, sensitized glioma cells to temozolomide by inducing apoptotic, rather than autophagic, cell death. Zanotto-Filio et al.40 suggested that autophagy has a protective role in gliomas as temozolomide/curcumin treatment in combination with 3-methyladenine (MA) leads to a reduction in cell viability.
The ability of cancer cells to sustain or intensify autophagic flux following irradiation is a key factor that defines resistance of glioblastoma cells to radiotherapy and temozolomide. Blocking this autophagy-mediated reparative cell process strongly sensitizes radioresistant gliomas. These findings may steer research towards the development of autophagy blocking therapies in combination with radiotherapy and temozolomide for treating glioblastoma, a highly lethal disease. An important finding raised by the current study is that radiation is dependent on dose and cell type for blocking autophagic flux, opening a new field of investigation for tailoring radiotherapy fractionation for each tumor and individual patient.
Footnotes
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received November 28, 2017.
- Accepted April 4, 2018.
- Copyright: © 2018, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.