Article Text

Abstract
Objective The stability of the covalently closed circular DNA (cccDNA) in nuclei of non-dividing hepatocytes represents a key determinant of HBV persistence. Contrarily, studies with animal hepadnaviruses indicated that hepatocyte turnover can reduce cccDNA loads but knowledge on the proliferative capacity of HBV-infected primary human hepatocytes (PHHs) in vivo and the fate of cccDNA in dividing PHHs is still lacking. This study aimed to determine the impact of human hepatocyte division on cccDNA stability in vivo.
Methods PHH proliferation was triggered by serially transplanting hepatocytes from HBV-infected humanised mice into naïve recipients. Cell proliferation and virological changes were assessed by quantitative PCR, immunofluorescence and RNA in situ hybridisation. Viral integrations were analysed by gel separation and deep sequencing.
Results PHH proliferation strongly reduced all infection markers, including cccDNA (median 2.4 log/PHH). Remarkably, cell division appeared to cause cccDNA dilution among daughter cells and intrahepatic cccDNA loss. Nevertheless, HBV survived in sporadic non-proliferating human hepatocytes, so that virological markers rebounded as hepatocyte expansion relented. This was due to reinfection of quiescent PHHs since treatment with the entry inhibitor myrcludex-B or nucleoside analogues blocked viral spread and intrahepatic cccDNA accumulation. Viral integrations were detected both in donors and recipient mice but did not appear to contribute to antigen production.
Conclusions We demonstrate that human hepatocyte division even without involvement of cytolytic mechanisms triggers substantial cccDNA loss. This process may be fundamental to resolve self-limiting acute infection and should be considered in future therapeutic interventions along with entry inhibition strategies.
- CHRONIC HEPATITIS
- HEPATITIS B
- ANTIVIRAL THERAPY
- BASIC SCIENCES
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
HBV chronic infection is guaranteed by the persistence of the viral genome forming a minichromosome, called covalently closed circular DNA (cccDNA), in hepatocyte nuclei.
HBV polymerase inhibitors do not target the cccDNA. Thus, HBV surface antigen seroconversion rates remain low and virus elimination is not achieved.
Cytolytic and cytokine-mediated mechanisms were shown to lower cccDNA loads. However, the fast recovery from acute self-limiting infection suggests that additional mechanisms may be involved to explain cccDNA clearance while the liver remains functional.
Experiments with HBV-related animal viruses and non-human hepatocytes indicated that cell division can lower cccDNA contents. However, knowledge about the capacity of nuclear cccDNA to survive human hepatocyte division is still lacking.
What are the new findings?
In vivo proliferation of HBV-infected human hepatocytes promotes strong cccDNA destabilisation and its clearance in the great majority of cells.
Nevertheless, the proliferative capacity of human hepatocytes displaying high HBV replication levels appears reduced in comparison to cells negative for HBV replication markers.
In the absence of immune-mediated cell killing, persistence of very few HBV-producing and apparently non-proliferating human hepatocytes serves as virus reservoir.
Treatment with HBV entry or polymerase inhibitors efficiently blocks reinfection of hepatocytes that have cleared cccDNA through cell division. Both treatments also hinder intrahepatic cccDNA accumulation.
How might it impact on clinical practice in the foreseeable future?
The strong cccDNA destabilisation caused by cell division suggests that curative therapeutic approaches should suppress HBV replication and involve controlled destruction of infected cells (ie, by boosting immune responses). This would accelerate cccDNA clearance also in surviving proliferating hepatocytes, while strategies aiming at preventing HBV entry and/or suppressing HBV replication would prevent reinfection of cured hepatocytes.
Introduction
Chronic infection with HBV (CHB) is characterised by the persistence of an episomal form of the viral genome, the covalently closed circular DNA (cccDNA), in the nuclei of infected hepatocytes.1 Although prophylactic HBV vaccines have been available for decades, the overall number of chronic infections remains high with >240 million individuals chronically infected worldwide who are at risk of developing liver cirrhosis and hepatocellular carcinoma.2 Therapies based on nucleoside analogues (NAs) effectively suppress HBV reverse transcription but do not target directly the cccDNA, whereas interferon treatment can induce immune clearance in only a minority of individuals.3–6 Thus, HBV surface antigen (HBsAg) seroconversion rates remain low and the infection typically relapses after treatment cessation. This is due to the refractory nature of the cccDNA in infected hepatocytes and to the inability to mount effective HBV-specific immune responses. The cccDNA, which originates from the incoming relaxed circular viral DNA (rcDNA), assembles in hepatocyte nuclei with histone and non-histone proteins to build a minichromosome that serves as template for the transcription of all viral RNAs.7 The pregenomic RNA (pgRNA) is an over-length complement of the viral cccDNA that is reverse transcribed by the viral polymerase within newly formed nucleocapsids, which are then enveloped and secreted in the bloodstream as progeny viruses.8 Unlike retroviruses, HBV does not require integration into the host genome for replication. However, integrated HBV DNA fragments are commonly found in infected patients9 ,10 and this phenomenon may contribute to carcinogenesis.2 Because the cccDNA is not directly targeted by NA treatment, prolonged therapy is needed to achieve significant cccDNA reduction in CHB.11–14 On the other hand, both cytopathic and non-cytopathic, cytokine-mediated mechanisms appear to contribute to cccDNA clearance during resolution of acute HBV infection, although their relative contributions, as well as the amount of hepatocyte destruction involved, are still debated.5 ,15–17 Moreover, the fast recovery from self-limiting acute infection suggests that additional mechanisms may be involved in cccDNA clearance while the liver remains functional. Immune cells such as CD8+ T cells and natural killer cells have the capacity to destroy the cccDNA together with the infected cell and to induce proliferation of neighbouring hepatocytes to compensate for cell loss.18 On cell division, the cccDNA molecules may be distributed among daughter cells leading to the dilution of the nuclear cccDNA pool. Since the cccDNA is not a cellular chromosome equipped with centromere structures, the HBV minichromosome may become distributed in an unequal way or even get lost during mitosis.17 Previous studies with patient liver biopsies,10 animal hepadnaviruses17 ,19 and mouse transplantation experiments involving Tupaia hepatocytes infected with the woolly monkey HBV (WM-HBV)20 pointed out an inverse relationship between hepatocyte turnover and cccDNA loads. However, knowledge on the proliferative capacity of HBV-infected human hepatocytes in vivo, as well as on the impact of mitosis on cccDNA activity and fragility is still scant. Moreover, an increasing body of evidence revealed unexpected differences between animal hepadnaviruses and HBV regarding their capacities to form cccDNA and control its pool size also by reimporting rcDNA-containing nucleocapsids from the cytoplasm.1 ,21–23 As all these factors will affect cccDNA persistence, we aimed to investigate the effect of human hepatocyte growth on HBV infection by employing human liver chimeric mice as a model permitting expansion of HBV-infected primary human hepatocytes (PHHs).
Materials and methods
Generation of humanised mice, infection, serial transplantation and drug administration
Homozygous uPA/SCID/beige mice (shortly termed USB) were housed and maintained under specific pathogen-free conditions according to institutional guidelines under authorised protocols as previously reported.4 To perform serial human hepatocyte transplantations, HBV-infected donor mice with high levels of human chimerism and viraemia were chosen for liver cell isolation by a two-step collagen perfusion and isolated cells were transplanted into naïve USB mice as described in the online supplementary methods. Before liver isolation, the caudate process of the mouse livers was removed to serve as a reference for future analyses (referred to as time point 0 or donor mouse). Some mice received subcutaneous injections of myrcludex-B (2 µg/g body weight) daily for 9 weeks,23 or lamivudine (Zeffix, GlaxoSmithKline, Brentford, UK) supplemented in the drinking water (20 mg/100 mL).
supplementary data
Serological and intrahepatic measurements
DNA and RNA were extracted from liver specimens using the Master Pure DNA purification kit (Epicentre, Madison, Wisconsin, USA) and the RNeasy RNA purification kit (Qiagen), respectively.4 Intrahepatic total viral loads were quantified with the help of primers and probes specific for total HBV DNA.24 Primers and probes specific for total HBV RNA and pgRNA24 were used for reverse transcription and amplification, while the expression of the human housekeeping gene GAPDH was used for normalisation. cccDNA levels were determined in whole cell liver DNA and in DNA samples extracted by Hirt, as well as after digestion with plasmid-safe ATP-dependent DNase (PSD), T5 exonuclease or a combination of Exonuclease I and III according to the conditions described in the online supplementary methods. All intrahepatic measurements were performed on three distinct liver pieces isolated per mouse. The detailed procedures used for all serological and intrahepatic measurements including virological assays, detection of viral integrations, gene expression analysis, mathematical calculations, immunofluorescence and RNA in situ hybridisation are provided in the online supplementary methods.
Statistics
Statistical analysis was performed with the GraphPad Prism V.5 software. The non-parametric Kruskal-Wallis test was used to compare the cccDNA decline in PHHs and the mouse liver. Dunn’s test for a group-wise comparison of the different time points was used as a post-test. The non-parametric Mann-Whitney U test was used to compare cccDNA before (d3) and during (d15, d30) proliferation in Hirt-extracted samples. The half-life for cccDNA decay was calculated on the linear regression of the log10 of each value determined over time. Significance of correlations was assessed using the F test. To assess whether the distribution of proliferating cells was equal among the HBV core antigen (HBcAg)-positive and negative PHHs, Fisher’s exact test (day 3) and χ2 test (day 14) was used. p Values <0.05 were considered to be statistically significant.
Results
HBV-infected human hepatocytes engraft and proliferate in recipient mice
To determine whether HBV-infected PHHs are able to reconstitute the livers of USB mice, we transplanted hepatocytes isolated from stably infected and highly viraemic humanised mice (median 2×109 HBV DNA copies/mL serum) in two independent experiments (figure 1A). Doing so, we made use of the intrinsic ability of the hepatocytes to integrate and grow in the liver of young uPA mice25 ,26 and assessed the proliferative capacities of engrafted PHHs in mice euthanised at different time points post-transplantation. Engrafted PHHs strongly proliferated as indicated by the expansion of human cell clusters over time (figure 1B and see online supplementary figure S1A), while no evidence of PHH death (<0.02% active caspase 3 positive cells) throughout the observation period was observed (online supplementary figure S1A, B).
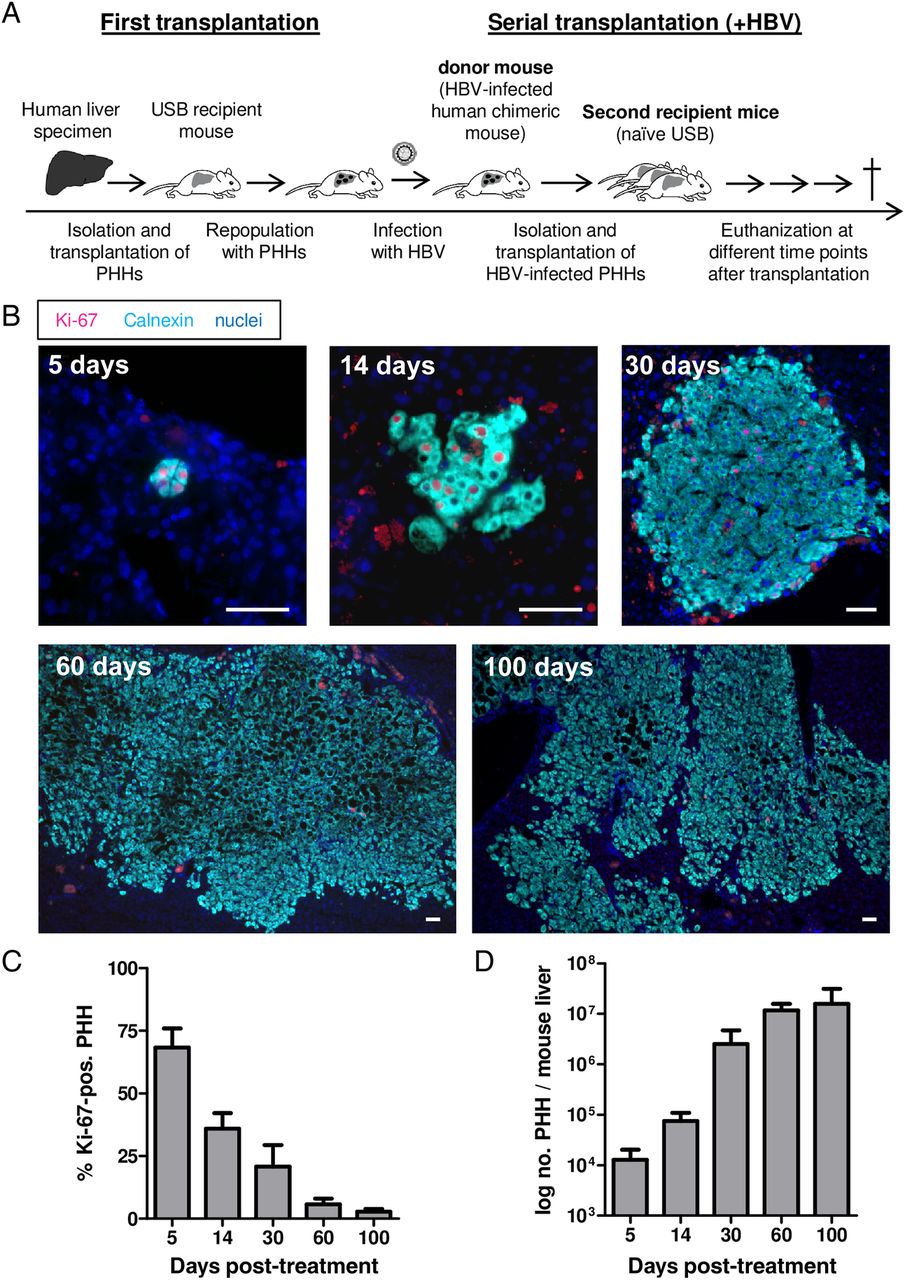
Experimental design and proliferation of HBV-infected primary human hepatocytes (PHHs) in recipient uPA/SCID/beige (USB) mice. (A) Schematic presentation of the experimental procedure used to produce chronically HBV-infected USB mice (first transplantation) and to induce proliferation of HBV-infected PHHs through another transplantation (serial transplantation). (B) Immunofluorescent staining of the proliferation marker Ki-67 (magenta) and calnexin (aqua) as a specific marker for human hepatocytes. Nuclei are stained blue with Hoechst. The panel shows one representative picture of a liver section for each time point post-transplantation as indicated in the upper left corner of each photograph. Scale bars=50 µm. (C) The proliferation index was set up by counting the percentage of Ki-67-positive PHHs on up to three sections per mouse and time point. (D) Mean amount of PHHs present in the mouse livers at different time points post-transplantation. PHH genome equivalents per ng liver DNA were determined by quantitative RT-PCR with primers specific for human haemoglobin β and results were scaled up to the whole liver mass at the time of sacrifice. Bars depict the mean of three to four mice and SD for each time point.
The proliferative stimulus was strongest at day 5 after transplantation (mean 68% Ki-67-positive PHHs) and progressively decreased in the following weeks (mean 21% at day 30, 3% at day 100) (figure 1C). To estimate the human repopulation index in mouse livers, human genome equivalents were quantified per ng liver DNA and scaled up to the liver weight determined at sacrifice.20 As depicted in figure 1D, we calculated a mean 3 log increase of PHHs per mouse liver between days 5 and 100 and an average of 7 cell doublings, thus confirming the engraftment and proliferation capacities of PHHs isolated from HBV-infected mice.
Steady-state levels of HBcAg decrease quickly on proliferation
Double staining for HBcAg and keratin 18 (human hepatocyte marker) showed that nearly all PHHs were HBcAg-positive in the donor mouse (figure 2A). Notably, most of the engrafted PHHs stained positive for HBcAg 3 days after transplantation (figure 2B). However, HBcAg staining decreased very rapidly and as soon as 2 weeks after transplantation only single PHHs stained HBcAg-positive (figure 2C), while HBcAg was almost completely lost at day 30 (figure 2D). At later time points, however, HBcAg staining slowly reappeared (day 60 and day 100 after transplantation, figure 2E, F), coinciding with the end of the proliferation phase in this mouse system. Complete reinfection required approximately 8 weeks, resembling closely the kinetics of a de novo infection in mice harbouring quiescent PHHs.23 To investigate whether there were differences in the proliferative capacities of cells clearly expressing HBcAg versus HBcAg-negative PHHs, we performed immunofluorescent triple-staining of HBcAg, Ki-67 and the human-specific cell marker SP100 at days 3 and 14 post-transplantation and quantified the respective fractions of HBcAg-positive, Ki-67-positive and double-positive PHHs. Representative pictures are shown in figure 2G, H. Three days post-transplantation 94% of counted PHHs appeared HBcAg-positive, while only 8% HBcAg-positive PHHs were found at day 14. Although both HBcAg-positive (28%) and HBcAg-negative (18%) PHHs stained Ki-67-positive at day 3, 2 weeks after transplantation, only 6% of HBcAg-expressing cells costained for Ki-67, while the percentage of proliferating PHHs within the HBcAg-negative fraction had remained similar (16%). This difference (p=0.018) accounted for a 2.6-fold reduced proliferative capacity of HBcAg-positive compared with HBcAg-negative PHHs at day 14. Notably, a few scattered HBcAg-positive and pgRNA-positive PHHs (<0.1%) were detected outside of human cell clusters at days 14 and 30 after transplantation (figure 2I, J). These cells, however, did not costain for Ki-67, suggesting that they had failed to proliferate.

Intrahepatic HBV core antigen (HBcAg) staining of HBV-infected primary human hepatocytes (PHHs) on proliferation in recipient mice. (A–F) Immunofluorescent staining of frozen liver sections for HBcAg (green), keratin 18 (red) and nuclei (blue). Keratin 18 was used to specifically stain human hepatocytes in the mouse livers. A representative picture of the donor mouse (A) and mice euthanised at different time points post-transplantation as indicated above the photographs (B–F) are shown. (G–H) Triple immunofluorescent staining of frozen liver sections for HBcAg (green), the proliferation marker Ki-67 (magenta), the human-specific marker SP100 (white) and nuclei (blue). Double positive PHHs for HBcAg and Ki-67 appear yellow when merged. (G) Depicts a typical situation at day 3; (H) Shows a typical situation at day 14 post-transplantation. For better visualisation, the clusters of human PHHs (=SP100-positive cells) are bordered by a broken white line. (I) Immunofluorescent staining for HBcAg (green), the human-specific marker keratin 18 (red) and nuclei (blue) of a mouse sacrificed 30 days after serial transplantation showing an example of one strongly HBcAg-positive PHH outside of a human cell cluster, which has lost HBcAg positivity in the course of proliferation. (J) RNA in situ hybridisation with probes specific for pregenomic HBV RNA (green dots) and human GAPDH transcripts (red dots) as a marker for human hepatocytes on a frozen liver section of a mouse sacrificed 30 days post-transplantation. Nuclei are stained blue with DAPI. This exemplary photograph shows a cluster of human hepatocytes (bordered by a broken white line) and a single double-positive human hepatocyte which produces pregenomic HBV RNA. Scale bars=50 µm.
Serological and intrahepatic viral parameters are reduced on proliferation
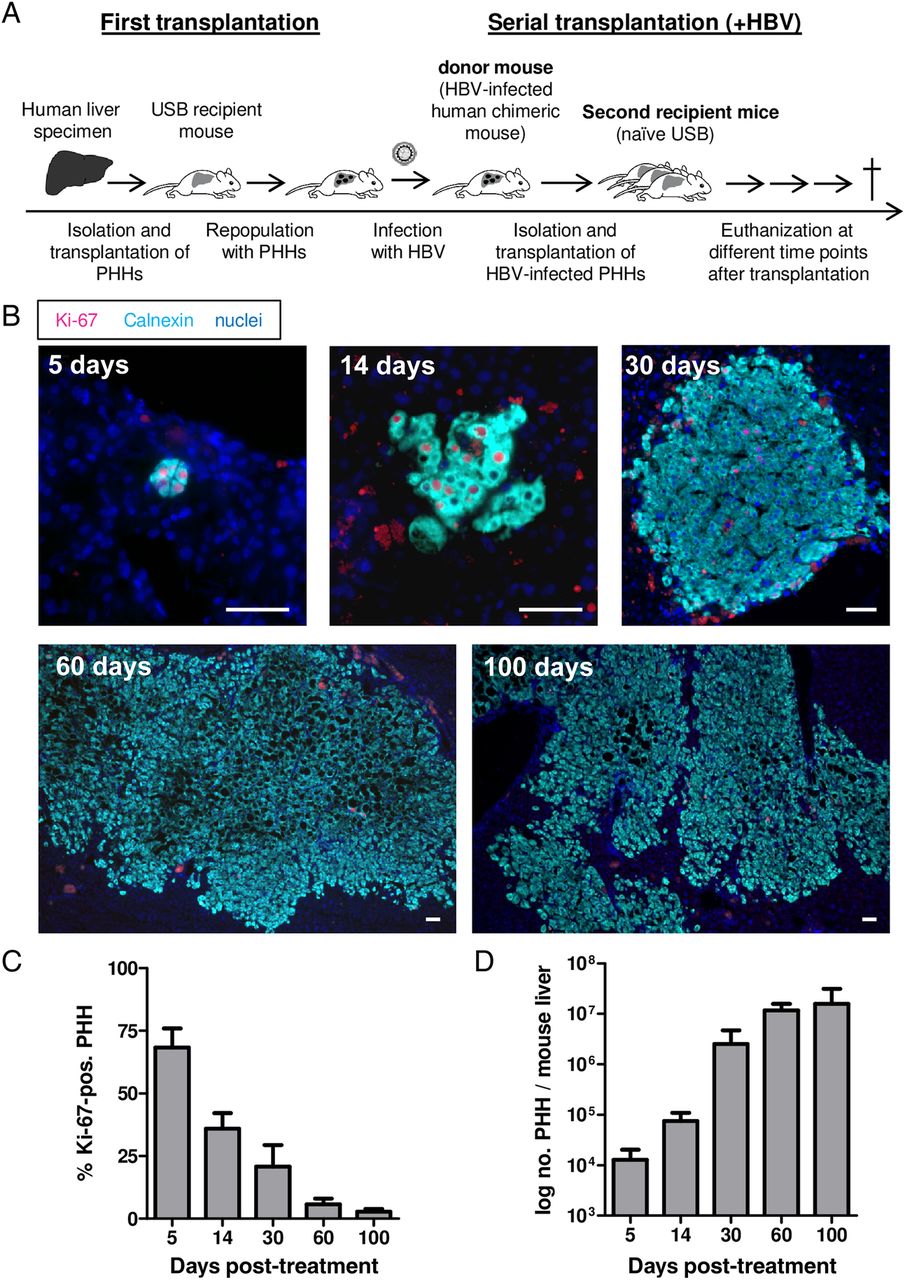
We further investigated the consequences of in vivo PHH proliferation on HBV replication. As shown in figure 3A, B, levels of circulating virions and HBsAg were low at early time points post-transplantation, reached minimum levels at day 30 but increased again thereafter. Accordingly, intrahepatic amounts of pgRNA (figure 3C) reached minimum expression levels at day 30 (Δ>2 log) and increased again at later time points. However, because pgRNA levels are normalised to the total amount of human cells and virus-free cells extremely increased in number over time, these measurements do not allow discriminating between a reduction of HBV RNA merely caused by amplification of virus-free hepatocytes or by the inhibition of viral replication in proliferating cells. In contrast, the ratio of pgRNA to cccDNA molecules did not change significantly over time (figure 3D), indicating that cccDNA transcriptional activity was not substantially affected in the milieu of liver regeneration. Most remarkably, cccDNA copies per PHH decreased dramatically during cell division (median 2.4 log reduction from day 3 post-transplantation until day 30; p=0.0047) (figure 3E). As depicted in figure 3F, when scaling up cccDNA contents to the liver mass at the time of sacrifice, we observed a significant reduction of the total cccDNA amounts per liver from day 3 or 5 until day 30 (0.7 log; p=0.0418 (Kruskal-Wallis test) or p=0.0314 (Mann-Whitney U test)). The optimised PSD and PCR protocol used here appeared to degrade most of the HBV non-cccDNA forms. However, an absolute assessment of the contribution of persisting rcDNA forms to the cccDNA signals is currently not possible. In view of these difficulties, we aimed to corroborate our findings by performing Hirt extraction to provide an independent procedure enabling enrichment of the cccDNA fraction and by using different nuclease digestion methods (T5 exonuclease or exonuclease I and III (personal communication Jianming Hu at the International HBV meeting 2016) to further clean up the cccDNA samples prior to PCR. As shown in online supplementary figure S2, all these methods yielded similar results both in terms of cccDNA reduction per cell and per liver (ie, Δ2.4 log cccDNA/PHH; p=0.0095 and 1.1 log cccDNA/liver; p=0.0095 using Hirt extraction and Mann-Whitney U test).

Changes of serological and intrahepatic viral markers induced by proliferation. (A–D) Viral markers of the donor mouse (=time point 0; as indicated by an asterisk) and the groups of recipient mice sacrificed at different time points post-transplantation performed in two independent experiments. Data points represent the median with range for 6–10 mice per time point. Viral titres are expressed as HBV DNA copies/mL serum (A), HBV surface antigen (HBsAg) as international units/mL serum (B) and pregenomic HBV RNA is normalised to the expression of the human housekeeping gene GAPDH (C). The amount of HBV RNA copies in (D) is normalised to the covalently closed circular (cccDNA) copy numbers present in the same liver piece. (E and F) Logarithmised values of cccDNA copies/primary human hepatocyte (PHH) (median log reduction 2.4 log, E) and cccDNA copies/mouse liver (median log reduction 0.7, F) from day 3 until day 30 of both experiments. Regression analysis was used to calculate the half-life of cccDNA amounts per human hepatocyte and mouse liver, respectively. Each dot represents a single mouse. The significance of the deviation from zero of the regression line is shown in the graph (F test).
By performing regression line analysis (see also online supplementary material, supplementary figure S2 and ref. 20), cccDNA half-life was calculated to be around 3 days if estimated per hepatocyte, regardless of the assay used, and ranged between 9 (after Hirt extraction) and 12 days (after PSD on total cell extracts) in the whole liver, suggesting that cccDNA was diluted among daughter cells and partially lost. However, linear regression analysis might be an oversimplification and a potential biphasic decay pattern of cccDNA reduction would be in line with the higher PHH proliferation rate determined in the first 2 weeks after transplantation. Nevertheless, viral markers reappeared as cell proliferation relented in all mice analysed from day 60 post-transplantation on (figure 3A–C).
To assess the possible contribution of the cytokine milieu in reducing cccDNA loads in these mice, we analysed the expression of human and murine inflammatory cytokines and growth factors that were reported to impact HBV replication or cccDNA stability (see online supplementary table 1).4 ,5 ,27–31 The expression of human genes was generally very low or under the detection limit, while the murine counterparts were easily detectable; however, neither murine nor human gene expression was specifically enhanced during the strong proliferation phase (between days 3 and 30) in comparison to values obtained at day 100 (see online supplementary figure S3). These results were also confirmed on the protein level using a magnetic Luminex screening assay for the human analytes tumour necrosis factor, interferon (IFN)B, IFNG, interleukin (IL)28A, IL28B, IL18, IL1B, IL6, epidermal growth factor and hepatocyte growth factor (data not shown), thus suggesting that these factors did not contribute substantially to the drop of cccDNA levels provoked by cell proliferation in this immunodeficient mouse model.
Recurrence of viral markers after the proliferation phase is a result of de novo infection
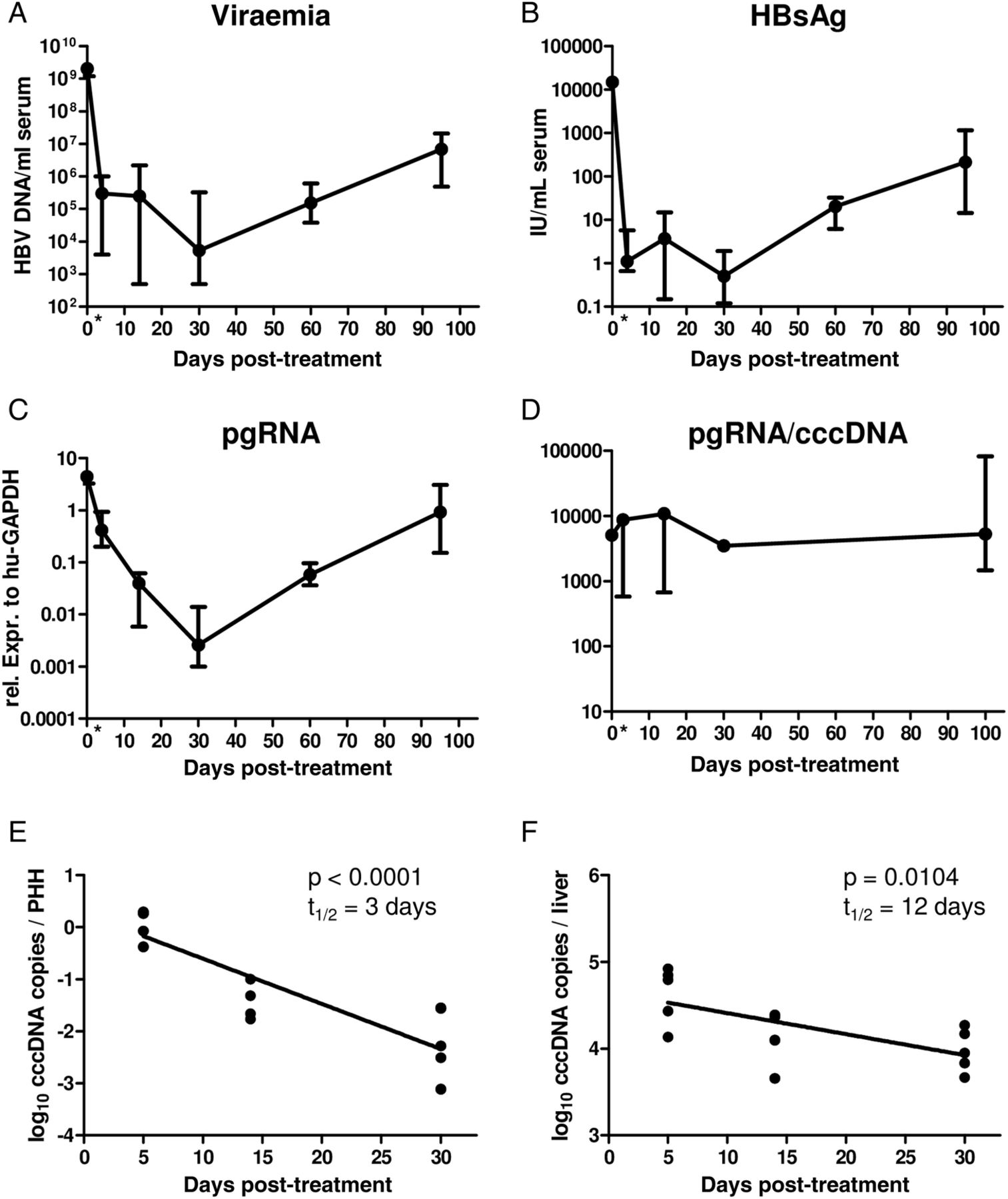
To test whether reappearance of HBV infection markers was due to de novo infection occurring after the proliferation phase had ended, we treated an additional group of mice with the viral entry inhibitor myrcludex-B.23 ,32 ,33 Starting at day 30 after transplantation, mice received the drug for 9 weeks. The lack of an increase in HBcAg-positive PHHs determined in treated mice (figure 4A) compared with untreated mice (figure 4B), together with the absence of an increase of intrahepatic cccDNA (figure 4C) and pgRNA (figure 4D) levels, demonstrated that prevention of HBV entry completely inhibited the recurrence of HBV infection in quiescent PHHs despite considerable levels of circulating virus (median 4×105 HBV DNA copies/mL). In a separate experiment, mice were treated with the NA lamivudine to keep viraemia levels low (median 4×104 HBV DNA copies/mL). Also in these animals viral rebound could be hindered as long as antiviral treatment was maintained (figure 4E, F). Altogether these results demonstrate that the reappearance of virological markers detected on completion of liver regeneration was mostly due to a de novo infection of quiescent PHHs, while cell division purged the cccDNA from the great majority of human hepatocytes. Notably, lamivudine treatment and myrcludex-B administration hindered the increase of intrahepatic cccDNA loads, suggesting that intracellular amplification of the cccDNA pool within already infected human hepatocytes barely took place.

Antiviral treatment blocks the recurrence of viral markers towards the end of the proliferation phase. (A and B) Immunofluorescence staining of liver sections stained for HBV core antigen (HBcAg) (green), keratin 18 (red) and nuclei (blue) of a mouse treated with myrcludex-B for 9 weeks (from day 30 to day 90 post-transplantation) (A) and an untreated animal (B) both sacrificed 90 days post serial transplantation. Scale bars=50 µm. (C and D) Comparison of viral markers in untreated animals (black line; experiment 2) and an additional group of five mice treated with the entry inhibitor myrcludex-B for 9 weeks (red line). (E and F) In an additional experiment, serially transplanted mice were treated with lamivudine from day 3 to day 100 and left untreated thereafter. For comparison, untreated animals (black line; experiment 2) are included. The graph in E shows covalently closed circular (cccDNA) copies/primary human hepatocyte (PHH) and F shows pregenomic HBV RNA normalised to the expression of the human housekeeping gene GAPDH. Curves depict median measurements with range of the donor mouse (time point 0) and recipient mice sacrificed after different time points post-transplantation.
Viral integrations are detected in donors and recipient mice but appear silent
To assess the occurrence of HBV integrations both in donor mice, which were infected after liver repopulation was accomplished, and in serially transplanted mice where HBV-infected PHHs had undergone multiple rounds of cell division, we performed gel electrophoresis to separate the low molecular weight fraction comprising rcDNA and cccDNA molecules from the high molecular weight fraction of genomic DNA possibly containing HBV DNA integrations prior to quantitative PCR (qPCR) analysis.20 As shown in online supplementary figure S4, all mice analysed—donor and recipient mice, including those treated with antivirals—showed the presence of HBV DNA sequences within the host genome, indicating that viral integrations occurred after proliferation of HBV-infected PHHs and on infection of quiescent PHHs.
To assess the expansion potential of these serially transplanted PHHs, we performed a second round of serial transplantation by isolating cells from one of the mice which had already been reconstituted with HBV-infected PHHs. Similar to the first serial transplantations, we observed a strong decrease of all viral markers until 30 days post-transplantation followed by viral rebound after the repopulation phase (see online supplementary figure S5A–D). Integration analysis revealed that the relative amount of integrated viral sequences varied among different liver specimens obtained from the same animal, suggesting that in some cases PHHs already harbouring HBV integrations had been expanded in recipient mice (see online supplementary figure S4C).
To characterise these HBV DNA integrations, we performed Alu-PCR followed by deep sequencing.34–36 Again, multiple HBV DNA integrations were identified in serially transplanted recipient mice and in donor mice that had been infected at a time when PHHs did not undergo proliferation. As shown in online supplementary figure S4D, HBV integrations included sequences corresponding to core, preS/S and X viral genomic regions. Concerning the integration site, most of HBV integrations were located at the level of repetitive or unidentified sequences, thus not allowing their precise characterisation. Nevertheless, single integration sites could be identified within or near coding regions of the human genome in five of the eight mice analysed by deep sequencing (summarised in online supplementary figure S4D).
The detection of viral integrations prompted us to investigate whether such sequences could substantially contribute to viral RNA and protein production and whether the strong expansion of HBV-carrying human hepatocytes could affect the differentiation status of the PHHs. After the second in vivo passage of PHHs, two out of three mice received lamivudine to hinder reinfection. Positive HBcAg and HBsAg staining could be determined only in the untreated mouse (figure 5A–D), where reinfection could take place after PHH repopulation was accomplished. As shown in figure 5E–H, simultaneous RNA in situ hybridisation with two probes recognising viral RNA sequences either in the pregenomic or the S and the X region revealed HBV RNA production in most PHHs in mice that underwent viral rebound, whereas the great majority of hepatocytes in treated mouse livers showed no sign of HBV RNA production. However, the very few HBV RNA-positive cells which were detected in treated mice (figure 5H) were producing both RNA species to similar extents resembling an active HBV infection. Accordingly, viraemia and HBsAg remained low in lamivudine-treated mice, while viral rebound exclusively occurred in the untreated mouse (figure 5I,J). The low levels of HBV RNA sequences and viral antigens in treated mice indicated that the contribution of HBV integrations to the production of viral antigens and/or truncated RNAs was negligible in our experimental setting. The detection of known liver cell differentiation markers such as HNF4A, KRT7, CTNNB1 and EPCAM both in donor and recipient mice also indicated that despite extensive proliferation of the human hepatocytes, no signs of hepatocellular dedifferentiation and cell transformation were observed (see online supplementary figure S6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Virological characterisation of HBV-infected primary human hepatocytes (PHHs) after two rounds of serial transplantation. (A and B) Immunofluorescent staining for HBV core antigen (HBcAg) (green), keratin 18 (red) and nuclei (blue) of frozen liver sections of mice sacrificed 100 days after the second transplantation, which were left untreated (A) or treated with lamivudine from day 30 until day 100 post-transplantation (B). (C and D) Immunofluorescent staining for HBV surface antigen (HBsAg) (green), keratin 18 (red) and nuclei (blue) of frozen liver sections of mice sacrificed 100 days after the second transplantation, which were left untreated (C) or treated with lamivudine from day 30 until day 100 post-transplantation (D). (E–H) RNA in situ hybridisation for pregenomic HBV RNA transcripts (green), S HBV RNA transcripts (red) and nuclei (blue) on frozen liver sections of mice sacrificed 100 days after the second transplantation. A scheme of the probe design (ie, the binding region and the number of ZZ pairs) is depicted in (E). A representative picture of a mouse which was left untreated is shown in (F) and one of a mouse treated with lamivudine from day 30 until day 100 post-transplantation is shown in (G). The photograph in (H), in contrast, represents a rare example of an HBV-positive human hepatocyte in a lamivudine-treated mouse producing pregenomic and S transcripts. Scale bars=50 µm. (I and J) Viral titres (I) and HBsAg (J) in the serum of the three recipient mice at 100 days after the second transplantation in HBV DNA copies/mL serum and international units/mL serum. Hatched bars indicate lamivudine treatment.
Discussion
The cccDNA is not affected directly by NA treatment and its longevity in non-dividing hepatocytes37 represents a key determinant of HBV persistence.1 Because the cccDNA is an extrachromosomal plasmid-like structure lacking centromeres, mitosis may accelerate its loss.38 ,39 By employing USB mice, we demonstrate that in vivo proliferation of HBV-infected human hepatocytes provokes a dramatic decrease of intrahepatic cccDNA contents. With an average cell doubling time of 5 days during the first 30 days after cell transplantation, the half-life of the cccDNA per human hepatocyte was calculated to be around 3 days, while the decrease of cccDNA within the whole liver appeared to progress with a half-life between 9 and 12 days depending on the assay used for cccDNA quantification. Although our optimised PSD protocol appeared to degrade most of the HBV non-cccDNA forms, cccDNA quantification by qPCR bare the drawbacks of the techniques available. Thus, to corroborate our findings, we included treatments with additional nucleases and an independent DNA extraction method (Hirt) enabling a selective enrichment of cccDNA molecules while removing significant amounts of genomic DNA and rcDNA. Even though Hirt extraction did not increase our capacity to retrieve cccDNA molecules, the specificity of the assay was clearly enhanced. Indeed, qPCR analysis using either primers spanning over the nick and gap (with higher affinity for cccDNA) or covering the S-region (measuring both rcDNA and cccDNA) revealed that the ratio of rcDNA to cccDNA was approximately 1, thus confirming that most rcDNA molecules could be removed. Notably, the magnitude of cccDNA decrease was confirmed regardless of the procedure used, although a smaller number of samples was still measureable after T5 or exonuclease I and III digestion possibly due to an overdigestion of cccDNA with these nucleases.
Altogether, these estimations provide the first direct in vivo evidence that proliferation of human hepatocytes significantly destabilises the HBV minichromosome even in the absence of immune-mediated cell killing or antiviral treatment. These findings support observations obtained from liver biopsies10 ,17 and are in line with studies employing HBV-related animal viruses.19 ,20 However, a less pronounced reduction of cccDNA was observed in duck HBV (DHBV)-infected growing ducklings,40 suggesting that the higher cccDNA copy number commonly determined in DHBV-infected hepatocytes and more efficient intracellular cccDNA replenishment via nuclear reimport of rcDNA from cytoplasmic capsids may have counteracted the reduction of the avian viral genome in proliferating duck hepatocytes. In this regard, cross-species transfection experiments highlighted fundamental differences between DHBV and HBV in their ability to generate the nuclear cccDNA pool. Both cccDNA formation and intracellular cccDNA amplification were shown to be less efficient in human cells compared with HBV-related viruses.21 In our study, treatment of mice with myrcludex-B proved that cell division led to the formation of cccDNA-free hepatocytes, efficiently prevented reinfection of quiescent cccDNA-cleared PHHs and even appeared to hinder detectable increase of intracellular cccDNA contents. Although such inefficient intracellular cccDNA amplification could be related to the model, the maintenance of a low cccDNA copy number per cell is in agreement with several studies involving human hepatocyte systems.11 ,12 ,22 ,41
Notably, cccDNA reduction appeared even stronger in mice treated with lamivudine compared with levels determined in myrcludex-treated mice (compare figure 4D with E). It is plausible that inhibition of reverse transcription further accelerated cccDNA decrease also by impeding infection of the reforming nucleus by rcDNA-containing capsids still present in the cytoplasm. Nevertheless, here we show that replenishment of the cccDNA pool either via import of rcDNA from the cytoplasm after cell division or via de novo infection through circulating virions could not compensate for the great cccDNA loss provoked by cell division. Consequently, the cccDNA could be efficiently purged from the great majority of PHHs.
Despite such strong cccDNA drop, complete viral clearance was not achieved indicating that a fraction of the cccDNA was refractory to further reduction. Accordingly, we estimated reduced proliferation capacities of HBcAg-positive compared with HBcAg-negative hepatocytes (2.6-fold) during liver regeneration. Because we solely relied on the expression of the core antigen for this analysis, we cannot exclude an underestimation of HBV-positive cells which had lost HBcAg expression but were still cccDNA-positive. However, the persistence of single non-proliferating cells clearly expressing viral proteins (HBcAg) at all time points also indicated that PHHs expressing high HBV levels may have growth disadvantages in comparison to uninfected hepatocytes. These results are in line with reports indicating lower proliferative capacities of HBV-replicating transgenic mice.42
Cytokines involved in anti-HBV immunity and liver regeneration were shown to inhibit HBV replication and to destabilise the cccDNA pool.4 ,5 ,27–31 Thus, cytokine induction could have contributed to the cccDNA loss determined in this study. However, we were unable to identify human or murine factors that were clearly enhanced during the phase of strong liver regeneration (from day 3 to day 30). These findings are not entirely surprising given that in these immunocompromised mice serum cytokine levels are generally low or undetectable even on HBV infection.24 Likewise, IL-1β and IL-18 levels were undetectable at all analysed time points, suggesting that neither cytoplasmic DNA activated the PHH inflammasome system nor danger signals appeared to be induced in the diseased liver parenchyma of young USB mice. In contrast to our previous study involving Tupaia hepatocytes and WM-HBV,20 cccDNA transcriptional activity did not appear affected by PHH division, since pgRNA amounts relative to cccDNA loads remained unchanged. Persistence of HBV replication in few scattered PHHs (<0.1%) was also demonstrated by RNA in situ hybridisation and was mirrored by a constantly low viraemia determined during the proliferation phase. From these results, we conclude that cccDNA reduction was mainly caused by cell division, whereas cytokines did not appear to contribute substantially cccDNA destabilisation.
Human studies indicated that clonal expansion of human hepatocytes may lead to the emergence of cells refractory to infection.10 The prompt viral rebound observed even after two successive rounds of cell transplantation demonstrated the great ability of quiescent human hepatocytes to maintain HBV infection susceptibility and hence their high differentiation status in vivo. Altogether, our experiments suggest that the reservoir for HBV reinfection lay within a few persistently infected PHHs that permitted intrahepatic viral spreading once cell proliferation had ended and in the absence of spreading inhibition strategies. Further characterisation of these cells and development of therapeutic strategies to specifically target such persisting HBV-producing cells may be fundamental to achieve viral elimination.43
Viral integrations were shown to occur at early steps of clonal tumour expansion and they are found in 90% of HBV-related hepatocellular carcinomas (reviewed in ref. 44). Moreover, cell culture and animal systems indicated that integrations of viral DNA sequences occur particularly in the presence of DNA damage.17 ,45 ,46 We identified fragmented viral sequences integrated within the host genome in PHHs that underwent extensive cell proliferation in the setting of HBV infection and in donor mice, where infection was established in quiescent PHHs that were not subjected to proliferative stimuli. Despite the presence of integrated fragments corresponding to core, preS/S and X regions of the HBV genome, neither the expression of HBV RNA sequences nor viral antigens (HBsAg and HBcAg) could be detected in liver sections of mice receiving antiviral treatment to prevent reinfection after liver reconstitution, thus indicating that such HBV DNA integrations were mostly silent or at least did not contribute considerably to the production of viraemia and circulating antigens (HBsAg) in this system. Nevertheless, during decades-long chronic infection in patients, emergence and expansion of hepatocytes harbouring HBV integrations may contribute to HBsAg production, as documented in HBV-derived hepatocellular carcinomas.44 ,47
The identification of cccDNA-negative cells containing ‘traces’ of the infection in form of integrations demonstrates—in line with previous studies48 ,49—that cccDNA can be cleared from human hepatocytes even in the absence of cell killing. However, cell death and regeneration are known to occur during resolution of self-limiting acute HBV infection1 ,45 ,50 and the fast recovery from acute self-limiting infection suggests that cytolytic and cytokine-mediated mechanisms are involved to explain cccDNA clearance in a liver which remains functional. It is plausible that both direct killing of infected cells and compensatory proliferation of HBV-infected hepatocytes play a key synergistic role in the process of clearing most of the intrahepatic cccDNA. In chronically HBV-infected patients, a transitory hepatic flare was shown to be beneficial to treatment outcome after stopping long-term NA therapy.51–53 According to this scenario, immune-mediated destruction of a proportion of infected hepatocytes accompanied by compensatory cell proliferation would accelerate the reduction of intrahepatic cccDNA loads and levels of circulating antigens, events that appear instrumental to gain immunological control. Owing to the limitations of this experimental immunodeficient model, monitoring of events occurring in chronically infected individuals will be essential. Although the high rate of cell division determined in this system is unlikely to apply for humans, killing of some infected cells and compensatory cell expansion may have significant effects on cccDNA levels and stability. On the other hand, cccDNA decrease may be even more pronounced if cell division occurs in immunocompetent systems under inflammatory conditions.
While more research is needed to develop therapeutic approaches boosting HBV-specific immune responses or agents directly targeting the cccDNA, our study reveals that cell division represents a natural Achilles heel in HBV persistence and underscores the importance of treatments protecting the hepatocytes from reinfection to maintain cccDNA clearance in cells which already resolved the infection.
Acknowledgments
We are grateful to Anne Groth and Roswitha Reusch for their excellent work with the mouse colony and to Gerlinde Apitzsch and Claudia Dettmer for their great technical help. We thank Davide Corti from Humabs BioMed for providing the HBV surface antigen-specific antibody.
References
Footnotes
Contributors MD, JP and ML initiated and supervised the study. MD, ML and LA designed experiments and analysed data. LA, TV and KG generated chimeric mice and performed animal experiments. LA performed hepatocyte isolations and molecular analyses. SU provided the myrcludex-B. TP and GR performed the Alu-PCR. CB performed the deep sequencing. JK performed the Luminex analysis. LA and MD wrote the manuscript. JP, ML, TV, TP, SU and AWL discussed the data and corrected the manuscript.
Funding The study was supported by the German Research Foundation (MD Heisenberg Professorship DA1063/3-2; Collaborative Research Centre SFB-841: A5). MD and SU also receive funding from the German Center for Infection Research (DZIF; TTU05.806 and TTU05.704).
Competing interests SU is co-applicant and inventor of patents protecting myrcludex-B.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Commentary