

Abstract
The ATM gene is mutated in the syndrome, ataxia-telangiectasia (AT), which is characterized by predisposition to cancer. Patients with AT have an elevated risk of breast and brain tumors Carrying mutations in ATM, patients with AT have an elevated risk of breast and brain tumors. An increased frequency of ATM mutations has also been reported in patients with breast and brain tumors; however, the magnitude of this risk remains uncertain. With the exception of a few common mutations, the spectrum of ATM alterations is heterogeneous in diverse populations, and appears to be remarkably dependent on the ethnicity of patients. This review aims to provide an easily accessible summary of common variants in different populations which could be useful in ATM screening programs. In addition, we have summarized previous research on ATM, including its molecular functions. We attempt to demonstrate the significance of ATM in exploration of breast and brain tumors and its potential as a therapeutic target.
keywords
Introduction
Alterations in the ATM gene are the main cause of the rare autosomal recessive disorder, ataxia-telangiectasia (AT). This condition is characterized by neuro-degeneration, cerebellar ataxia, oculomotor apraxia, choreoathetosis, telangiectasias of the conjunctivae, frequent infections, immunodeficiency, radiosensitivity, and a predisposition to cancer, and was described as a specific disease entity in 1957. AT occurs early in childhood, and its frequency varies from 1 in 40, 000 to 300, 000 births in various countries and ethnic groups1,2. To date more than 400 disease-related mutations have been identified in ATM, approximately 70% of which result in the production of truncated protein3,4 (http://www.vmresearch.org/atm.htm). The majority of mutations are unique and they are uniformly distributed throughout the open reading frame of the gene, without any hotspots. Despite the 100% penetrance of truncating mutations, the low penetrance of missense mutations often leads to an insignificant phenotype5. The prevalence of ATM mutations ranges from 0.5% to 1% in Western populations1,6; however, homozygosity for the same mutation is rare.
Maintaining genomic integrity and stability is essential for all types of cells with the critical function of inhibiting tumor development, and ATM is a key factor in these processes. ATM has various functions, including a key role in the recognition and repair of double-strand breaks (DSBs). The response to DNA damage includes recognition of damaged DNA, recruitment of repair proteins, signaling to cell cycle checkpoints, and facilitation of apoptosis via regulation and activation. Since ATM is involved in many of these processes, it could be considered a master regulator of the DNA damage response7-9.
In the brain, the processes involved in maintaining genomic stability and integrity are particularly important, since neurons are terminally differentiated and unable to divide. Therefore, they have evolved multiple overlapping mechanisms for DNA damage repair10. Consistent with the significance of DNA repair to the health and survival of brain cells, mutations in DNA repair genes often cause syndromes which include pronounced defects in the central nervous system (CNS)11-13. AT, caused by mutations in the ataxia-telangiectasia gene, is characterized by progressive degeneration of the cerebellar cortex, and sometimes brain tumors14, particularly medulloblastomas and gliomas15-19. Cytogenetic and molecular deletions of chromosome 11q (the ATM gene region) in medulloblastomas suggest the presence of one or more tumor suppressor genes in this region, which could have important roles in brain tumors pathogenesis20-23. Therefore, there appear to be two different pathways that contribute to the AT disease process.
Radiation exposure is associated with an elevated risk of breast cancer24; therefore, the function of ATM makes it a plausible candidate for a role in breast cancer predisposition. Indeed, an association between AT and breast cancer was first reported by Swift et al.25, before the ATM gene was cloned, when they identified an excess of breast cancers in the relatives of individuals with AT.
ATM structure
The ATM gene was mapped by genetic linkage analysis to chromosome 11q22–23 in 198826 and was identified by positional cloning in 199527. The gene contains 66 exons, of which 62 encode a 350-kDa protein of 3056 amino acid residues. The ATM gene covers 160 kb of genomic DNA, and is transcribed in a broad range of tissues to produce an mRNA of approximately 13 kb with a coding sequence of 9168 bp. Exons 1a or 1b are differentially spliced in alternative transcripts, the initiation codon lies within exon 4, and the final 3.8 kb exon contains approximately 3.6 kb of untranslated sequence at the 3' end28. ATM is a member of the family of phosphatidylinositol-3-kinase (PI3K)-related protein kinases (PIKK), has serine–threonine protein kinase activity, and includes a PI3K-like domain at its C terminus that comprises almost 10% of the protein. Other important functional domains of ATM are the leucine zipper, which has sequence homology to the Saccharomyces pombe Rad3 protein, and a proline-rich region29-31.
ATM function
ATM is the major initiator of a signaling cascade that responds to DSBs, and which operates not only in humans, but also other eukaryotes (Table 1). Normally, ATM is present in cells in the form of an inactive dimer or multimer complexes. Following DNA damage, ATM undergoes autophosphorylation at S1981 leading to the separation of the inactive complex to form highly active monomers. Subsequently, through activation of signaling pathways, and via phosphorylation of numerous substrates, DNA repair can take place32, during which two central responses to DNA damage, including the activation of cell-cycle checkpoints and the initiation of DNA repair, are facilitated.
ATM roles in different species
Cell-cycle checkpoint function
ATM links the recognition of DNA damage to the maintenance of genomic integrity and stability by activating checkpoints that lead to a delay in the progress of cells carrying damaged DNA through the cell cycle. The progression of the cell cycle is controlled by cyclin-dependent kinase (CDK) complexes (Figure 1). Sequential events in the cell cycle rely on checkpoints to guarantee their step by step progression. Also, if the DNA is damaged, checkpoints arrest the cell cycle by delaying the activity of CDKs. ATM is involved in the regulation of these checkpoints after exposure to ionizing radiation (IR)44-48.
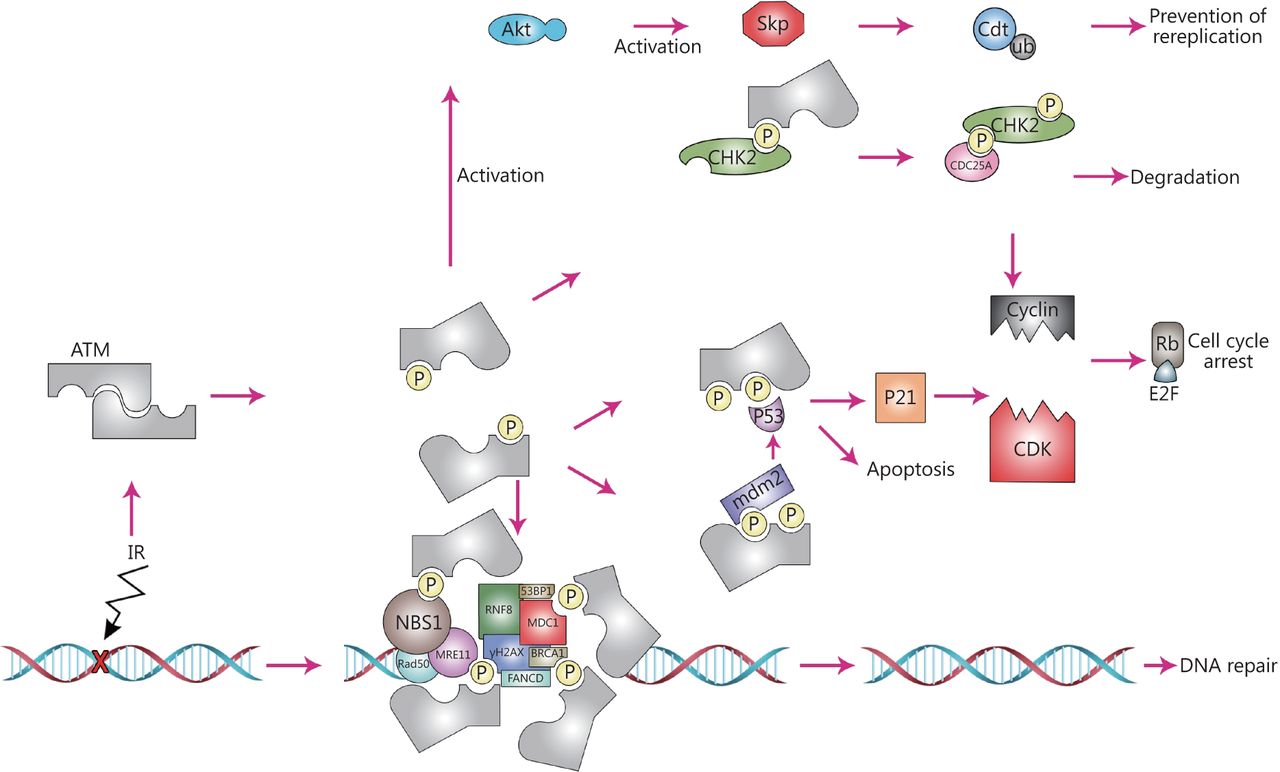
ATM plays multiple roles in human cell biology. To be highly active, ATM disunites to monomers; like other PI3Ks for activation it needs a stimulus and leads to phosphorylation of several different substrates. On the other hand, the activity of ATM can be regulated via some of these substrates. Indeed, they provide a platform for ATM. In the area of damaged DNA, ATM takes part in the phosphorylation of the histone variant H2AX, resulting in production of γH2AX. ATM then phosphorylates the adapter protein MDC1. These phosphorylation events form a docking station for many elements involved in the DNA damage repair system, including the RING-finger ubiquitin ligase, RNF8 that binds to phosphorylated MDC1. RNF8 then leads to ubiquitation of γH2AX, resulting in its stabilization. Stabilized γH2AX is the required platform for recruitment of p53 binding protein 1 (53 BP1) and BRCA1. Following their recruitment, 53BP1 and BRCA1 get phosphorylated by ATM, activating DNA repair processes. ATM also interacts with the MRN complex elements, such as MRE11, NBS1 and Rad50, which bind to double strand DNA and act together as a sensor of DNA damage. This interaction leads to an effective response to DNA damage. In the next levels of ATM dependent mechanisms, p53 transactivates its target genes including CDK inhibitor p21, resulting in the inhibition of the Cyclin-CDK complex formation and hindering G1 to S phase transition. mdm2 which negatively regulates p53, is also phosphorylated by ATM, resulting in abrogation of its interacting potential and stabilization of p53. If occurred in S-phase, DNA DSBs triggers CHK2 activation (phosphorylation on threonine 68) by ATM, leading to the phosphorylation of CDC25A, causing its degradation. Genomic stability is also regulated by ATM where it activates Akt, triggering active Skp that in turn regulates the critical replication licensing factor, Cdt. This process is vital for the maintenance of genome by licensing only one replication event in any given region.

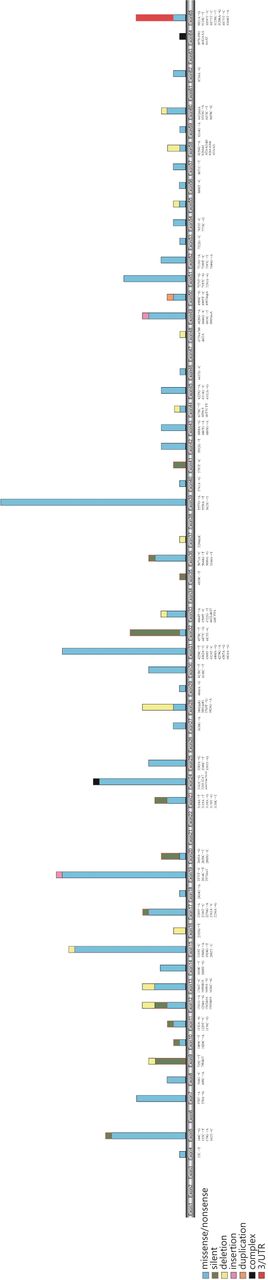
Mutations of ATM gene in exonic region according to their repetition in different studies in breast cancer patients.
Loss of p53 function, which can occur because of mutations in tumor cells, abrogates cell cycle G1/S checkpoints49. DNA damage leads to a rapid induction of p53. The demonstration of delayed and reduced induction of p53 in AT cells following IR exposure was the first link between ATM and p5350,51. In normal cells, the expression level of p53 is maintained by Mdm2, an E3 ubiquitin ligase52,53, which prompts p53 ubiquitination and degradation. In cells harboring DNA damage, ATM phosphorylates p53 and Mdm2 on amino acids Ser15 and Ser395, respectively54,55. This leads to nuclear accumulation of p53 in response to IR, as a result of reduced Mdm2 activity and consequently inhibits shuttling of p53 from the nucleus to the cytoplasm for polyubiquitination and proteasomal degradation. Following DNA damage, in addition to p53 activation and stabilization via modifications56, ATM also inhibits its nuclear export and the ability of Mdm2 to degrade p53, which is an important step in p53 activation54,57,58. Accordingly, the expression of p53 and ATM has been identified as correlated in several cancer cell types59.
In brain tumors, p53 risk and protective haplotypes are associated with glioblastoma and ATM in meningioma60; however, Kheirollahi et al.61 reported no correlation of expression between ATM and p53 in grade I and II astrocytomas and meningiomas. Accordingly, Barros et al.62 argued that ATM likely regulates gene expression in an indirect way. It was also suggested by Kheirollahi et al.61, that various complex mechanisms could contribute to this interaction in brain tumors. In breast cancer, however, Angèle et al.63 showed a correlation between the expression of p53 and ATM. It has also been demonstrated that patients with tumors containing concomitant p53 mutations and low ATM expression levels exhibit inferior survival rates, compared with those with wild-type p53 and high ATM expression levels64. Concannon et al.65 suggested that a number of ATM alleles exhibit anti-neoplastic effects, which might be caused by changes in the activity of ATM as a DNA damage initiator or a p53 regulator in breast tumors.
There are two pathways that enforce the G1-S checkpoint; the main and delayed response is p53-dependent. Activation, accumulation, and stabilization of p53 leads to transcriptional regulation of a set of genes associated with the cell-cycle. Among those is the CDK inhibitor, p21, which controls the binding of CDK2 to Cyclin D. Binding and activation of CDK2/Cyclin D eventually results in G1/S arrest, a process that requires inactivation of Rb, following progression from G1 to S-phase66 (Figure 1). A study investigating astrocytoma and meningioma brain tumors, revealed that associations between ATM, Cyclin D2, and Rb expression are disordered in higher grade tumors, while in lower grade tumors, different patterns of correlation were observed61. In lower grade meningiomas and astrocytomas, the expression levels of CCND2 (Cyclin D2), P53, Rb1, and ATM were significantly positively correlated. ATM has been identified as expressed at higher levels in high grade brain tumors, possibly because it is up-regulated in response to DNA damage in higher grade malignancies. The correlation between expression of genes has been demonstrated in various different grades of tumor, indicating a non-specific pattern, even for genes that interact directly61. In meningioma tumor cells, ATM and p53 protein levels were found to be low and very low, respectively. In contrast, astrocytoma tumors present a mixture of low and high ATM and p53 protein levels. The existence of tumor cells with high ATM and p53 expression levels is considered a reliable indirect sign of benign status in meningiomas. These observations are comparable with the high expression levels of p53 and ATM proteins in healthy individuals. Interestingly, low levels of ATM protein expression, together with clones of cells with low levels of Rb protein, indicate a harmonic expression pattern in astrocytoma tumors67.
ATM also prevents entry to S-phase in response to DNA damage, via a second, p53-independent, pathway. CHK2 is a key target of ATM in this pathway, and is activated in response to DNA damage through phosphorylation on various sites, including the Thr68 regulatory residue68,69. Activation of CHK2 results in phosphorylation of its target, CDC25A, stimulating its degradation. Reduction of CDC25A blocks entry to S-phase, as it prevents dephosphorylation and activation of CDK270. Mehdipour et al.71 showed that CDC25A is up-regulated in breast tumors and associated with poor survival. These authors suggested that CDC25A is a pivotal prognostic cell cycle marker that could be used for diagnosis of high risk breast cancer. High expression levels of CDC25A have also been observed in glioma brain tumors72.
As mentioned above, CDC25A phosphorylation by ATM and CHK2 also controls the intra-S phase checkpoint. In addition, in response to DNA damage, ATM is involved in phosphorylation of multiple factors, providing sufficient time for cells to repair DNA lesions during the intra-S phase checkpoint. In this context, BRCA1, FANCD2, NBS1, and SMC1 are phosphorylated by ATM on residues Ser-1387, -222, -343, and -957 (or -966), respectively73.
The process that prevents cells containing damaged DNA from entering mitosis is the G2-M checkpoint, which is regulated via several parallel mechanisms. Phosphorylation of BRCA1 on Ser1423 by ATM, inhibits the formation of the CDC2/Cyclin B complex74. In addition, BRCA1 catalyzes CtIP ubiquitination, and its subsequent interaction with chromatin leads to G2-M checkpoint control75. P53 dependent G2 arrest, CDC25C phosphorylation via ATM, and its subsequent events, are other key mechanisms that are induced in response to DNA damage during the G2-M checkpoint73.
DNA repair function
As one of the first lines of protection following DNA damage, ATM is rapidly recruited to sites of DSBs (Figure 1), accompanied by the MRN complex (MRE11, NBS1, Rad50). The latter acts as a sensor of damaged DNA, and ATM is not required for its localization76. In contrast, the MRN complex binds to double stranded DNA and is necessary for the response to DNA damage, which depends on the complete activation of the ATM77,78, providing a platform and acting as substrate for ATM79,80. The endo- and exonuclease activities of ATM are important for stimulation of ATM by the MRN complex81. MRE11 leads to production of small DNA fragments which can stimulate ATM activation at the site of DNA DSBs82; nevertheless, Lee et al.83 noted that the nuclease activity of MRE11 in the MRN complex is not crucial for ATM activation. In addition, ATM directly interacts with the MRN complex through the C-terminus of NBS1 (also known NBN)84. All these molecular interactions at the site of DNA DSBs result in an effective DNA damage response that can eventually lead to DNA repair. Awasthi et al.81 proposed that the activation of ATM may be associated with its dephosphorylation. Upon its recruitment, ATM induces the phosphorylation of Ser139 of H2AX, resulting in production of γH2AX85, which in turn provides a platform for binding of the adapter protein, MDC1, and its phosphorylation by ATM86,87. Although dispensable for the initial recruitment of ATM to the site of DNA DSBs, MDC1 is necessary for its retention. Formation of phosphorylated γH2AX-MDC1 complexes at the site of DSBs provides a docking platform for proteins involved in DNA repair and related signaling pathways, including the RING-finger ubiquitin ligases, RNF168 and RNF888-91. RNF8 recruits p53 binding protein 1 (53BP1) and BRCA1 as a result of ubiquitination of γH2AX, resulting in its stabilization. Moreover, 53BP1 and BRCA1 are phosphorylated by ATM92. Thus, phosphorylation events mediated by ATM are essential for the repair of DNA DSBs (Figure 1).
Other roles of ATM
Another important role of ATM is its involvement in the cellular response to oxidation state. Following oxidative stress, changes to the integrity of several intermolecular disulfide bonds lead to the induction of ATM. At DNA DSBs, ATM activation is through phosphorylation of Ser1981, which results in dimer dissociation. In contrast, under oxidative conditions, disulfide bonds between the dimer subunits form covalent linkages, resulting in activation of its kinase activity; this function is independent of Ser1981 phosphorylation93,94.
ATM also prevents re-replication by regulation of Ctd1 stability, and acts as a guardian of genomic integrity and stability during unperturbed cell-cycle progression. Interestingly, depletion of ATM alters the level of Cdt1, which is a critical replication licensing factor (along with p27 in some cells), without changing the expression of Cyclin D1 or p21, factors regulated by Skp2. This results in strict regulation of Cdt1, ensuring that only one replication program occurs. Of several already unanswered questions, “how does ATM regulate Akt activation?” is notable, as it could hold the key to understanding ATM regulated signaling pathways involved in cell growth and inhibition of apoptosis95,96 (Figure 1).
ATM variants in breast cancer
Northern Europe
The most frequent variants of the ATM gene in patients from northern Europe (Finland, Denmark, and the Netherlands) include 2572T>C, 3161C>G, 5558A>T, 2119T>C, 4258C>T, and 5557G>A. The 2572T>C variant, is a missense variant causing Phe858Leu amino acid change in exon 19; another variant at this site, 2572insT, was reported in the Netherlands. Two of these variants, 3161C>G (Pro1054Arg) and 5558A>T (p.Asp1853Val), encode amino acid substitutions predicted to have deleterious effects on protein structure. Gutierrez-Enriquez et al.107 showed that lymphoblastoid cell lines expressing the ATM variant, 3161C>G, exhibited chromosomal radiosensitivityin vitro, possible due to a dominant-negative effect on ATM function170. The alterations, 2119T>C and 4258C>T, are missense variants in exons 15 and 31, respectively, encoding Ser707Pro and Leu1420Phe protein changes. Heikkinen et al.97 proposed that the 5557G>AATM variant modifies cancer risk when present in cis with the IVS38-8T>C mutation, which is associated with bilateral breast cancer in patients of Finnish origin. They also suggested that such composite alleles lead to lowATM expression levels that may influence mis-splicing of exon 39 (Table 2).
ATM variants have been reported in the breast cancer patients from northern Europe
Southern, central, and western Europe
The most frequent variants of the ATM gene in patients from southern, central and western Europe (Switzerland, Britain, Germany, France, and Spain) are 2572T>C and 3161C>G. The 3161C>G variant was not reported in the Polish population, and another frequent variant, 2572T>C, did not vary significantly between cases and controls in the Polish study115. In addition, the 2572T>C variant was not reported in Italian population studies (Table 3) .
ATM variants have been reported in the breast cancer patients from southern, central and western Europe
Eastern Europe
The most important and frequent variant of the ATM gene in eastern Europe is the nonsense mutation, 5932G>T, which leads to inclusion of a termination of translation at codon at position 1978. It has been reported in the Czech Republic, Austria, Belarus, Ukraine, and Russia. Interestingly, it has rarely been reported in other European countries, other than Poland. Thus, it appears that this variant may have arisen exclusively in eastern Europe. Bogdanova and colleagues, reported a five-fold higher frequency of this mutation in breast cancer patients compared with controls, indicating a role in predisposition to breast cancer susceptibility for the E1978X amino acid change encoded by this allele. This mutation appears to be remarkably frequent in patients from eastern European countries, including Russia, Belarus, and the Ukraine, whereas its incidence is somewhat lower in Poland114 (Table 4).
ATM variants have been reported in the breast cancer patients from eastern Europe
America
The most frequent variants of the ATM gene in the American breast cancer patients, including 5558A>T (D1853V) and 5557G>A (D1853N), which have been reported in all different regions and ethnicities in America; although, 5558A>T was not reported in Japanese and Mexican Americans. Bretsky et al.121 noted that 5557G>A is the most important polymorphism, while other missense variants are rare, and do not appear to be overrepresented among breast cancer patients compared with controls in Americans (Table 5).
ATM variants have been reported in the breast cancer patients in different regions and ethnic groups of America and Canada
Australia
The most important missense variant of the ATM gene in Australian breast cancer patients is 7271T>G. This is a missense variant causes a valine to glycine substitution at position 2424 of the ATM protein (p.Val2424Gly), and has no influence on any recognized functional domain, suggesting that it is deleterious. This variant has also been reported in the USA, Canada and Britain, which are countries with high immigrant populations. Goldgar and his colleagues stated that women with theATM 7271T>G variant are at high risk and that screening for this variant in cancer-prone families, even those lacking changes inBRCA1 or BRCA2, is essential for management and genetic counselling130. Another prevalent variant in Australian breast cancer patients is the 3802delG truncating mutation in exon 28. IVS10-6T>G is a further important variant, which has been reported in all regions of the world. This variant leads to incorrect splicing of exon 11, producing ATM mRNA and protein molecules of less than 10% of the full-length versions144. Although multiple studies suggest that ATM IVS10-6T>G contributes to an elevated risk of breast cancer105,131, a large meta-analysis revealed it is not correlated with increased risk of breast cancer in the overall study population145. The role of the IVS10-6T>G mutation in breast cancer development is suggested to both be determined by its functional consequences and mediated by family history and BRCA1 and BRCA2 mutation status101,119.
Asia
Mehdipour and colleagues141 proposed that the polymorphism 5557G>A (located in exon 39 ofATM) could be considered a predisposing factor for the development of familial breast cancer. In this study, Iranian women with breast cancers were divided into two subgroups. The first group were selected at random and the second group had a positive family history; both were compared with a group of healthy controls. The study revealed a carrier frequency of 31% in the disease group, compared with 18.6% in controls. In the randomly selected group, the carrier frequency was 12.5%, whereas the rate was 26.9% in subjects with a family history of breast cancer141. To address the clinical significance of this polymorphism, a meta-analysis study by Gao and colleagues146 concluded that there was no association between the 5557G>A polymorphism and disease. In contrast, Tapia and colleagues126 found a clear positive association between this polymorphism and a high risk of bilateral breast cancer development. They analyzed the frequency of 5557G>A and the intronic variant, IVS24-9delT, individually or in combination, and found a frequency of 20.3% heterozygosity among patients and 7.5% in controls, while only 1% homozygosity was detected in both groups. Furthermore, this variant was described in European countries by Angéle and colleagues110. The authors found that homozygotes were more frequent among patients with breast cancer who had received radiotherapy. This finding led to the proposal of the association of this variant with elevated radiosensitivity in breast tissue, and the suggestion that this alteration is a predisposing factor for reaction effects occurring after radiotherapy110. In another study, González-Hormazábal and colleagues125 found three ATM polymorphism, IVS24-9delT, IVS38-8T>C, and 5557G>A (the latter more common, at 20.6%), among Chilean patients with familial breast cancer, who were negative for mutations in BRCA1/2. They suggest that these three alterations, either alone or in combination, together with environmental factors, could increase the risk of breast cancer, as a result of raised chance of genetic instability or failure of the DNA damage response. Youlden et al.147 reported variable mortality rates among different regions, with huge increases in some Asian countries compared with low rates in Australia. Although all breast cancer patients with ATM mutations, who had inferior survival rates and died during the follow-up period, carried the 5557G>A variant, this was not a statistically significant finding148 (Table 6).
ATM variants have been reported in the breast cancer patients in Asia, Australia and New Zealand
ATM variants in brain tumors
To date, 739 mutations have been identified in the ATM gene (HGMD 2015). Patients with AT and CNS involvement and patients with CNS tumors can have ATM gene disruption. In response to DNA damage, requirement for special mechanism in terminally differentiated cells and also localization of ATM at a key biochemical node10 indicate strong evidence of a link between ATM and CNS; however, adequate studies are lacking. To date, the evidence for the influence of specific ATM mutations and polymorphisms in different types of brain tumor is limited (Table 7, Figure 3). The first study of the association between ATM variants and brain tumors was performed by Liberzon and colleagues14, who screened 13 medulloblastomas and found that the only altered ATM sequences were the known polymorphisms, F858L and D1853N, which were present in five patients (38%). The reported frequency of the D1853N polymorphism was consistent with that in a previously published study of patients with medulloblastoma149. Moreover, D1853N has been reported in the majority of studies65, 109, 127, 150, 151, and is considered the most important fundamental alteration identified by genotyping of the ATM gene in patients with brain tumors152.

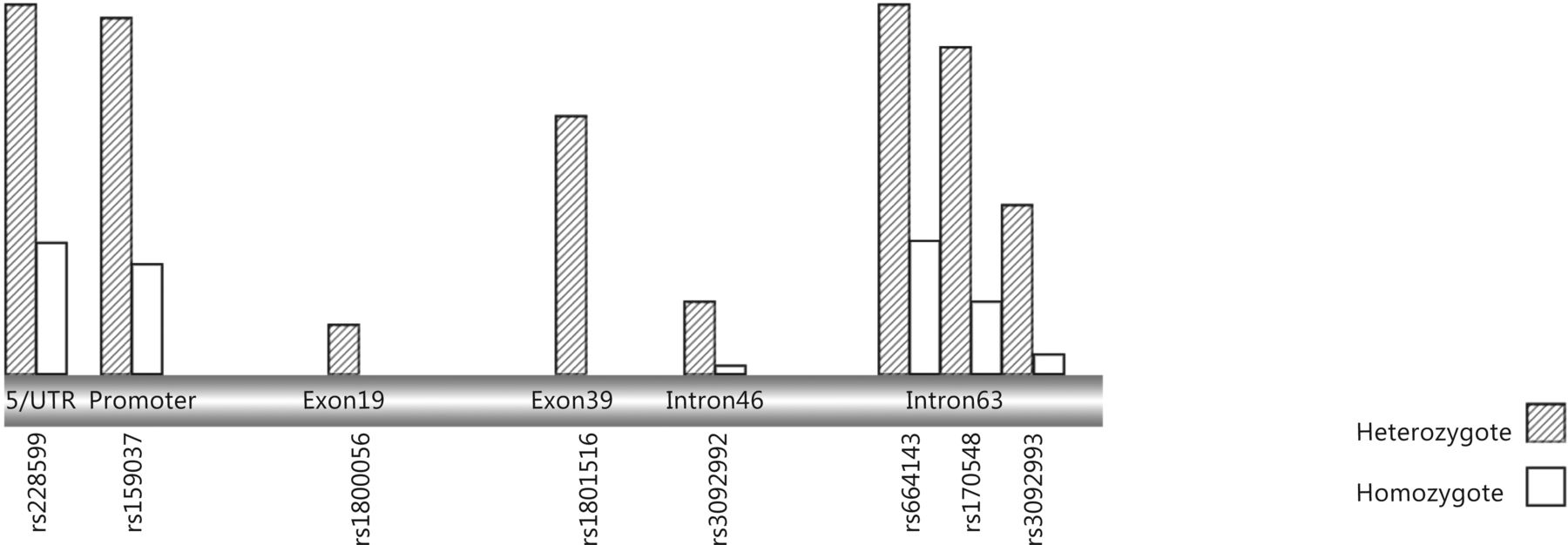
ATM gene variants in brain tumors.
In glioma, four variants have been reported, with 4578C>T the only exonic polymorphism identified in a patient population from the north of the UK153. This variant was also detected in patients with breast cancer from Finland, Denmark, Germany, France, Spain, Poland, and the USA, and at the amino acid level encodes Pro1526Pro (exon 30). Zhao et al.154 identified a novel polymorphism in the ATM promoter region in gliomas. This variant, 111G>A, is located in the non-coding region of theATM gene and has no direct influence on the amino acid sequence of the ATM protein; however, it may effect splicing, modification, or RNA stability, and thereby the mode of expression of the ATM protein155. Bioinformatics analysis to explore the potential mechanism of ATM mRNA expression regulation by this polymorphism suggested that, ATM sequences containing the SNP, rs189037, were transcriptionally repressed by AP-2a156. To date, the ATM protein kinase has been referred to as a DNA damage sensor and a therapeutic target in tumors157.
ATM variants have been reported in different types of brain tumors
Breast to brain metastases
Brain metastases are the most common form of intracranial spread of tumors, accounting for 15%–40% of patients with metastatic conditions160-162, whereas 42% of metastatic breast tumors are solitary163. Overall, breast and lung cancers are the most likely to progress to brain metastases164. Patients with breast cancer are remarkably likely to develop brain metastases, leading to a disease state with undesirable consequences for quality of life and more complicated responses to therapy. The incidence of brain metastases varies between 140, 000 and 170, 000 cases per year, and in recent years has risen, seemingly because of the prolonged survival of patients with primary tumors who have undergone aggressive treatment162,165.
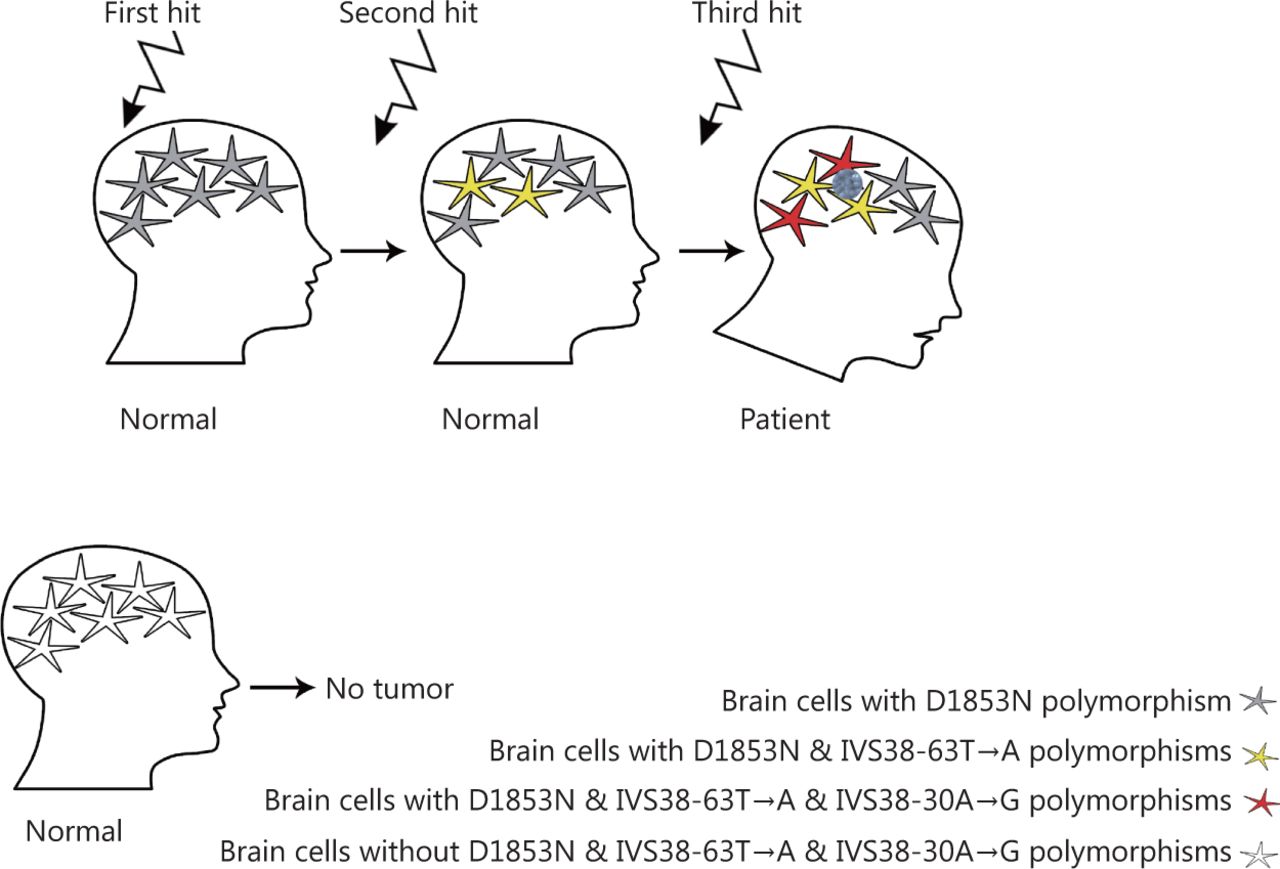
Gachechiladze et al.166 reported a significant correlation between shorter survival and phosphorylation of BRCA1 in patients with brain metastases of lung cancer. The role of ATM in BRCA1 phosphorylation and the interaction of these proteins is established166,167. In addition, variants of ATM and BRCA1 have functional effects in breast cancer141,168. Thus, ATM may have a crucial role in tumors involving breast-brain metastases. Studies investigating ATM variants in this type of breast tumor will shed new light on this field; however, no such study has been published to date. Recently, the D1853N mutation in ATM was reported in tumors with ovarian to brain metastases, and ATM and BRCA1 were found to be the most commonly altered genes169. Mehdipour et al.141 explored the role of D1853N in disease susceptibility among breast cancer patients and healthy individuals, and found this variant is a risk factor for metastatic progression. Furthermore, metastatic progression has been classified as a stepwise molecular alteration process of cancer development which requires three hits, D1853N, IVS38-63T>A, and IVS38-30A>G, occurring sequentially in astrocytes152 (Figure 4). In addition, Mehdipour et al.67 found a triangular correlation between methylation of the ATM promoter and protein expression of ATM with the D1853N variant. Evaluation of the D1853N variant alongside other alterations in patients with breast cancer, particularly those with breast to brain metastases, could lead to development of appropriate clinical management approaches.

Three hit hypothesis. Two novel heterozygously intronic alteration, i.e., IVS 38- 63 T>A and IVS 38-30 A> was found in proband located within 3 regions of splicing site. Two non-inherited events including IVS38-63T>A and IVS38-30A>G, resulting from two separate courses of evolution in proband occurred on the same chromosome which was different from the inherited D1853N polymorphism (data is adopted from Mehdipour, et al. 2008).
Heikkinen and colleagues97 proposed a cancer risk-modifying consequence of the IVS38-8T>C alteration occurringin cis, as it was reported as associated with bilateral breast cancer in patients of Finnish origin. They suggested that the function of such a composite allele leads to low ATM expression levels, that may be related to the mis-splicing of a region of exon 39. These data emphasize the importance of evaluation of ATM expression, in addition to exploration of its variants. The only study to do this was that of Salhia et al.170 who demonstrated that ATM gene expression was commonly down-regulated in patients with breast cancer and brain metastases. Combined functional assays, including copy number variation and gene expression analyses, demonstrated the down-regulation of ATM, revealed cell cycle and G2-M pathway enrichment, and have potential to lead to innovative therapeutic strategies. These findings are of particular importance, since they suggest rare therapeutic options for tumors involving breast to brain metastases.
Therapeutic opportunities
Given its broad range of functions, in processes from cell cycle regulation to DNA repair, ATM represents a therapeutic target molecule in tumors of the brain and breast. The discovery and introduction of anticancer drugs targeting molecules involved in the cell-cycle and checkpoint regulation, such as cell-cycle or checkpoint inhibitors171, can also influence ATM indirectly. The majority of cancer therapy studies related to ATM have focused on its significance in radiotherapy172-175. Molecular mechanisms that cause resistance to radiotherapy in glioma cells have been linked to the expression of ATM, in cooperation with other pro-survival networks176. A key mechanism leading to inactivation of tumor suppressor genes is methylation of their promoters. The methylation pattern of the ATM promoter has been investigated in a few studies of different types of brain tumors177,178. For example, Mehdipour et al.67 found that more than 73% of brain tumors exhibit methylation of the ATM promoter. A strong correlation between ATM promoter methylation and its protein expression has also been established. In another study, in which three glioblastoma cell lines (U87, T98G, and U118) were analyzed, methylation of the ATM promoter was only found in the T98G cell line178; however, despite its normal methylation status, the level of ATM protein was decreased in U87 and U118 glioma cells, which also exhibited elevated sensitivity to radiation. The use of siRNA to silence ATM expression can increase tumor cell radiosensitivity, as reported recently175. In brain metastases from the majority of epithelial cancers, the blood brain barrier (BBB) limits the use of chemotherapy as the first-line treatment option, with radiotherapy considered most effective. However, resistance to radiotherapy leads to disease recurrence and therapeutic failure. Yang et al.179 reported increased radiosensitivity in response to treatment with the CHK1 inhibitor, AZD7762, in lung cancer cell lines and a xenograft model. In lung cancer cell lines, the mechanism underlying this phenomenon was identified as the interaction of AZD7762 with ATR/ATM-mediated CHK1 phosphorylation, stabilization of CDC25A, and suppression of cyclin expression. In addition, in a lung cancer xenograft model of brain metastases, the median survival period was enhanced by AZD7762179.
Tumors with DNA repair pathway deficiency are sensitized to platinum drugs that induce such double strand breaks180. Hence, breast cancer patients with BRCA1 mutations exhibit increased responses to treatment with cisplatin181. Tumors with ATM mutations are also highly sensitive and responsive to platinum chemotherapy182.
Recently, Fann et al.183 evaluated the small molecule, NSC745887, in glioblastoma cells and found that it caused high expression of γH2AX, leading to DNA fragmentation, enhanced G2-M arrest, and apoptosis via induction of DNA damage responses.
Future perspective
Adequately informative gene expression/polymorphism profiles of human brain tumors are not available, because of disease heterogeneity and lack of comprehensive studies. Thus, comprehensive understanding of tumorigenic processes requires additional complementary investigations and pedigree-based analyses. Hence, more studies are required to fully address the etiology of brain tumors and differences in ATM variations, particularly in brain metastases. Nevertheless, monitoring of ATM alterations in brain tumors is a rather limited approach that has been restricted to a few types of brain tumors. Some reports of evaluation of ATM gene expression are available in different tumors, particularly brain tumors. Neurons are unable to enter the cell cycle; however, cancerous brain cells lose this feature and reactivate cycling, indicating that their templates of activation or inactivation differ fundamentally to normal ones. Accordingly, there does not appear to be a specific template for the correlation of the expression pattern of key factors in the DNA repair process, even among genes known to closely interact, including p53, ATM, Rb and Cyclin D. Studying the mRNA expression level of the above-mentioned genes in human brain tumors is necessary. To clarify any relationship between ATM functions and its variants, complementary research studies are required. The newly discovered function of ATM in response to oxidative conditions blurs any clear distinction between the DNA repair process and other cellular functions of ATM.
According to available data on breast tumors, it could be concluded that ATM gene status, is highly variable depending on the available data on the patient’s population origin and pedigree. In addition, a primary obstacle to effective therapy for brain tumors, specifically higher grade tumors, is rooted in resistance to radiotherapy. The remarkable resistance of cells lacking ATM to radiation is a key finding worthy of consideration. What are the expectations of the clinicians and scientists fighting to eradicate breast cancer and brain tumors? Comprehensive insights into the molecular involvement of the ATM gene have the potential to facilitate more reliable clinical management, including improvement of patient survival.
Footnotes
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received January 31, 2018.
- Accepted April 16, 2018.
- Copyright: © 2018, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.
- 9.↵
- 10.↵
- 11.↵
- 12.
- 13.↵
- 14.↵
- 15.↵
- 16.
- 17.
- 18.
- 19.↵
- 20.↵
- 21.
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.
- 31.↵
- 32.↵
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- 44.↵
- 45.
- 46.
- 47.
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.
- 90.
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.
- 99.
- 100.
- 101.↵
- 102.
- 103.
- 104.
- 105.↵
- 106.
- 107.↵
- 108.
- 109.↵
- 110.↵
- 111.
- 112.
- 113.
- 114.↵
- 115.↵
- 116.
- 117.
- 118.
- 119.↵
- 120.
- 121.↵
- 122.
- 123.
- 124.
- 125.↵
- 126.↵
- 127.↵
- 128.
- 129.
- 130.↵
- 131.↵
- 132.
- 133.
- 134.
- 135.
- 136.
- 137.
- 138.
- 139.
- 140.
- 141.↵
- 142.
- 143.
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.
- 159.
- 160.↵
- 161.
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.
- 174.
- 175.↵
- 176.↵
- 177.↵
- 178.↵
- 179.↵
- 180.↵
- 181.↵
- 182.↵
- 183.↵
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.