Article Text

Abstract
Objectives Patients with gallbladder carcinoma (GBC) lack effective treatment methods largely due to the inadequacy of both molecular characterisation and potential therapeutic targets. We previously uncovered a spectrum of genomic alterations and identified recurrent mutations in the ErbB pathway in GBC. Here, we aimed to study recurrent mutations of genes and pathways in a larger cohort of patients with GBC and investigate the potential mechanisms and clinical significance of these mutations.
Design We performed whole-exome sequencing (WES) in 157 patients with GBC. Functional experiments were applied in GBC cell lines to explore the oncogenic roles of ERBB2/ERBB3 hotspot mutations, their correlation with PD-L1 expression and the underlying mechanisms. ERBB inhibitors and a PD-L1 blocker were used to evaluate the anticancer activities in co-culture systems in vitro and in vivo.
Results WES identified ERBB2 and ERBB3 mutations at a frequency of 7%–8% in the expanded cohort, and patients with ERBB2/ERBB3 mutations exhibited poorer prognoses. A set of in vitro and in vivo experiments revealed increased proliferation/migration on ERBB2/ERBB3 mutation. Ectopic expression of ERBB2/ERBB3 mutants upregulated PD-L1 expression in GBC cells, effectively suppressed normal T-cell-mediated cytotoxicity in vitro through activation of the PI3K/Akt signalling pathway and contributed to the growth and progression of GBC in vivo. Treatment with an ERBB2/ERBB3 inhibitor or a PD-L1 monoclonal antibody reversed these immunosuppressive effects, and combined therapy revealed promising therapeutic activities.
Conclusions ERBB2/ERBB3 mutations may serve as useful biomarkers in identifying patients who are sensitive to ERBB2/ERBB3 inhibitors and PD-L1 monoclonal antibody treatment.
Trial registration number NCT02442414;Pre-results.
- gallbladder carcinoma
- whole-exome sequencing
- ERBB2/ERBB3
- Programmed death-ligand 1(PD-L1)
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Uncovering the genomic alterations may provide potential therapeutic targets for patients with gallbladder carcinoma (GBC).
A preliminary study with 32 patients has identified highly frequent mutations of ErbB signalling pathway.
What are the new findings?
High-frequency ERBB2/ERBB3 mutations were identified in 157 patients with GBC and was confirmed to be associated with worse overall survival.
Activated ERBB2/ERBB3 mutations promote tumour cell growth and migration, as well as upregulate PD-L1 expression to induce immune evasion of GBC cells in vitro and in CD34+ humanised NCG mice in vivo.
PD-L1 blockade can enhance the efficacy of anti-ERBB therapy in GBC cells carrying mutant ERBB2/ERBB3.
How might it impact on clinical practice in the foreseeable future?
ERBB2, ERBB3 and other factors involved in the downstreaming signalling pathways might be potential therapeutic targets for patients with GBC.
ERBB2/ERBB3 mutations could be served as biomarkers to identify patients who are sensitive to PD-L1 therapy.
Introduction
Oncogenesis is a multistep process that involves a number of genetic and epigenetic alterations. Growing evidence suggests that genomic alterations acquired during oncogenesis help tumour cells escape immune surveillance.1 Gallbladder carcinoma (GBC) accounts for 80%–95% of biliary tract cancers and is associated with poor prognosis and a median survival of less than 1 year,2 in part because most patients are diagnosed at advanced stages.3–5 Little is known about how GBC occurs or why the tumour cells are highly metastatic.
Genomic instability and mutability are likely to play vital roles in GBC development, given that these factors participate in tumourigenesis and tumour progression in many other cancers.6 7 Next-generation sequencing and profiling have identified many genomic mutations in various genes, including TP53 and ERBB3, in patients with GBC.8 9 In our previous study of 32 patients with GBC, we identified several high-frequency mutations in genes encoding proteins participating in canonical signalling pathways, including 15 genes in the ErbB pathway.10 To extend this work, the current study performed whole-exome sequencing (WES) to identify novel somatic mutations important in gallbladder carcinogenesis in a larger number of patients. We detected high-frequency mutations in TP53 (27%), KMT2C (11%) and SMAD4 (11%) and oncogenic mutations in ERBB2 (7%) and ERBB3 (8%). We found that the ERBB2/ERBB3 mutations promoted proliferation and migration of GBC cells in vitro and in vivo, which may reveal a mechanism explaining poor patient prognosis. Further, ERBB2/ERBB3 mutations stimulated PD-L1 expression in GBC cells, which led to the immune suppression of T cells targeting GBC tumour cells. These results suggest that ERBB2/ERBB3 mutations promote PD-L1-mediated immune escape in GBC.
Materials and methods
Patients
The current study included 157 patients with pathologically verified GBC who underwent cholecystectomy from 2012 to 2016 at Xinhua Hospital, Zhongshan Hospital or Eastern Hepatobiliary Surgery Hospital. All of these hospitals are located in Shanghai, China. Clinical staging was performed based on the seventh staging system of the American Joint Committee on Cancer. Clinicopathological features of the patients are presented in online supplementary tables 1 and 2. Pathological verified tumour tissue samples were processed in parallel for WES, and the adjacent gallbladder tissues that did not contain cancer cells were recognised as the normal controls. The respective ethics committees of the participating hospitals approved the study protocol, and all patients provided written informed consent.
Supplementary file 1
Whole-exome sequencing
Genomic DNA was extracted using the QIAamp DNA kit (Qiagen, Valencia, CA, USA). A Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA) and a Thermo NanoDrop 2000 (Thermo, Wilmington, DE, USA) were used to determine DNA concentration and quality. An Agilent 2100 Bioanalyzer (Thermo) was used to determine DNA length and integrity. The library for WES was created using the standardised protocol recommended by Illumina, and whole-exome enrichment was performed using the SeqCap EZ capture kit (Roche). VARSCAN software was used to identify somatic single-nucleotide variations and indels. MutsigCV software and the MUSIC suite were used to analyse the significance of identified mutations and predict potential pathways affected, respectively. More detailed information about WES is described in the online supplementary methods.
Supplementary file 2
Cell culture and reagents
The human GBC cell lines GBC-SD and NOZ, the genome-wide mutation status of which is described in online supplementary tables 3 and 4, were cultured as previously described.10 Primary antibodies against ERBB2 (#2165), ERBB3 (#12708), FLAG tag (#14793), PD-L1 (#13684) and CD8 (#70306) were obtained from Cell Signalling Technology (Beverly, MA, USA). Primary antibodies against CD3 (ab16669) and CD4 (ab133616) were obtained from Abcam (Cambridge, UK). The ERBB2/ERBB3 inhibitor sapitinib (AZD8931), ERK1/2 inhibitor (SCH772984), AKT1/2/3 inhibitor (MK-2206 2HCL) and anti-PD-L1 antibody atezolizumab (MPDL3280A) were purchased from Selleckchem (Shanghai, China).
Knockdown and overexpression of ERBB2/ERBB3 mutants
Somatic ERBB2 and ERBB3 mutations were introduced into a pCMV6 vector carrying the ERBB2/ERBB3 open reading frame encoding a C-terminal fusion with the DDK tag as previously described.10 Transgene expression was verified using cell lysates in Western blotting for ERBB2/ERBB3. In addition, mutant and wild-type ERBB2/ERBB3 genes were cloned into pCDH lentiviral expression vectors, which were transfected into GBC cells to establish stable cell lines.
Cell viability, clonogenicity and transwell migration
These assays were performed as described in the online supplementary informations.
Fluorescent in situ hybridisation (FISH) assay, immunohistochemistry, western blotting, immunofluorescence and quantitative RT-PCR
The assays were performed as described in the online supplementary methods.
Microarray analysis
The assay was performed as described in the online supplementary methods.
PD-L1 detection on the cell surface
Cells were suspended in 100 µL of cell staining buffer and incubated at room temperature for 30 min with phycoerythrin (PE)-conjugated antihuman PD-L1 antibody (#557924, BD Biosciences). After washing in staining buffer, stained cells were subjected to fluorescence-activated cell sorting (BD Biosciences), and the results were further analysed using FlowJo V.7.6.1 software.
T-cell killing assay, co-culture and expression of IFN-γ; and IL-2
The assays were performed as described in the online supplementary methods.
Sapitinib and atezolizumab treatment assay in vitro
GBC cells were seeded into 96-well plates (2×103 cells/well), incubated overnight, co-cultured with peripheral blood mononuclear cells (PBMCs) and treated with vehicle, atezolizumab (10 µg/mL), sapitinib (100 nM) or both atezolizumab and sapitinib. After treatment for 48 hours, we removed the culture supernatant, including suspended PBMCs and tumour cells, and washed the attached tumour cells thrice with culture medium.
Mouse xenograft models and toxicity assay in vivo
Female Balb/c nude mice (6–8 weeks old, 18–20 g) were purchased from the Shanghai Laboratory Animal Centre of the Chinese Academy of Sciences (Shanghai, China). CD34+ humanized NCG mice (NOD-Prkdc em26Cd52 Il2rg em26Cd22/NjuCrl), which are described in the online supplementary material and methods, were purchased from the Nanjing Biomedical Research Institute of Nanjing University. All mice were housed under specific pathogen-free conditions following the guidelines of the Ethics Committee of Xinhua Hospital of the School of Medicine at Shanghai Jiaotong University. NOZ cells (1×106) expressing the ERBB2S310Y mutant and the ERBB3V104L mutant were injected subcutaneously into the right flank of nude mice. On day 3 after inoculation, mice were injected intraperitoneally with 100 mg/kg sapitinib or vehicle daily. In addition, mice were injected intraperitoneally every 4 days with 10 mg/kg atezolizumab or human IgG1 control (BE0297; Bio X Cell). Tumour volumes were measured every 3 days using a digital calliper and the following formula: volume=π/6×length×width2.2Body weight was measured every 5 days.
Statistical analyses
Genome MUSIC software was used to identify relationships between the mutation status of genes and signalling pathways and the overall survival of GBC patients. Potential associations between mutations and clinical characteristics were assessed for significance using the χ2 test or Fisher’s exact test in the case of small cell counts. Univariate survival analysis was performed using the Kaplan-Meier test. Associations between risk factors and survival were assessed using multivariable Cox proportional-hazards models to calculate HRs and 95% CIs. Intergroup differences in cell viability and migration were assessed for significance using the two-tailed Student’s t-test based on the results of at least three independent experiments. Statistical analyses were performed using SPSS V.18.0. Results were considered statistically significant when the two-sided p value was <0.05.
Results
WES to identify genomic mutation signatures and frequencies in GBC
To gain insight into the genetic alterations associated with the pathogenesis of GBC, we performed WES in a cohort of 125 patients. WES achieved 92.0-fold coverage for GBC and 81.0-fold coverage for adjacent non-cancerous tissue (online supplementary figure 1). These data were combined with the results of the 32 patients from our previous study10 to identify disease-associated and prognosis-associated somatic mutations (online supplementary tables 1 and 2). Annotation analysis using Variant Effect Predictor11 yielded 17 684 non-silent coding mutations (112.6 per sample; online supplementary tables 5 and 6). The median number of mutations was 37.0 per sample, which is significantly lower than in the TCGA landmark cohorts (online supplementary figure 2). Transition mutations were significantly more frequent than transversions (online supplementary figure 3A,B).
Supplementary file 3
Supplementary file 4
Supplementary file 5
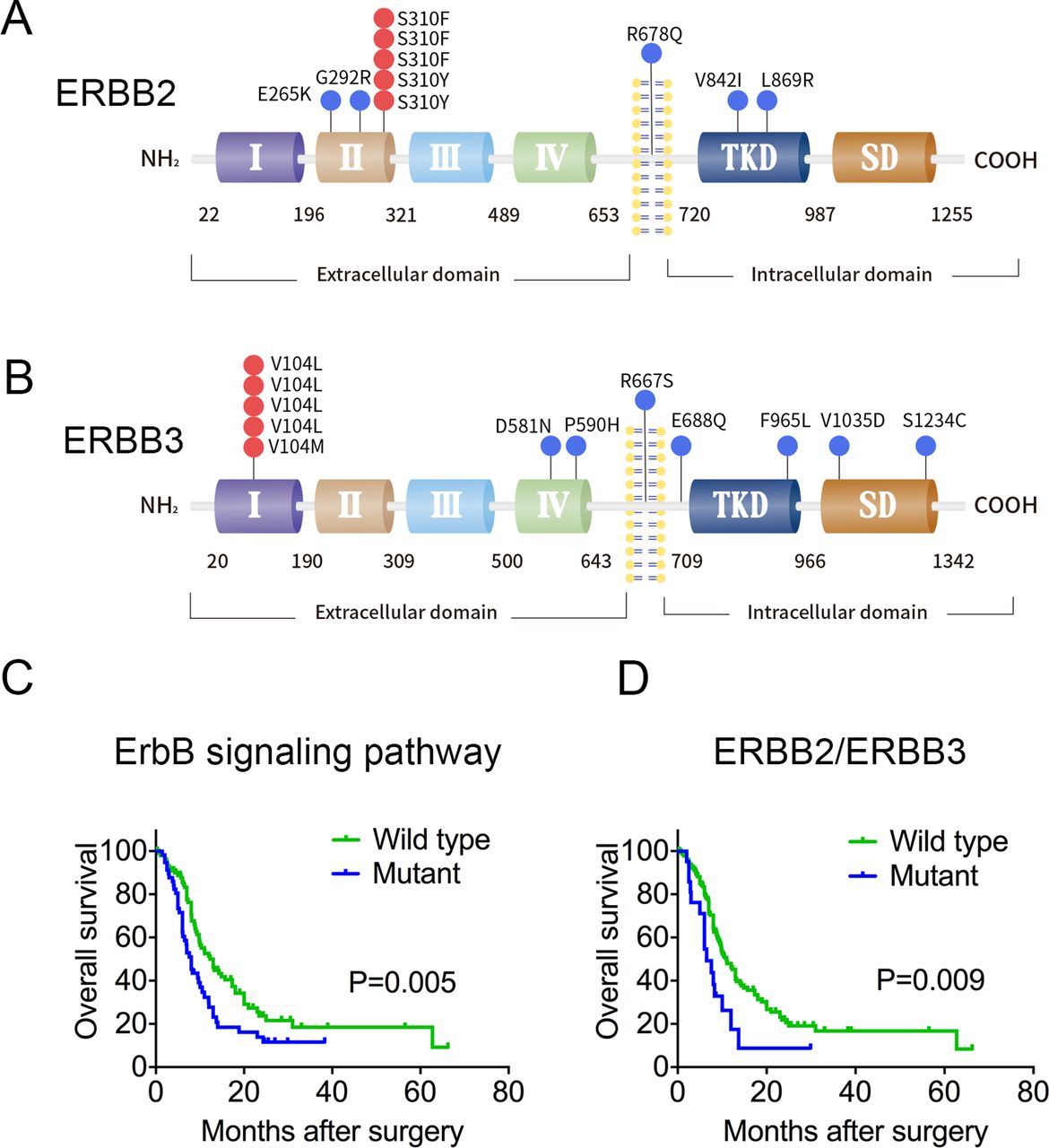
Somatic mutations in ERBB2 and ERBB3 and their associations with GBC patient prognosis. (A, B) Somatic missense mutations in ERBB2 and ERBB3 are indicated at the top of the corresponding domains in the protein. Red dots indicate hotspot mutations; blue dots indicate other mutations. SD, C-terminal signalling domain; TKD, tyrosine kinase domain. (C) Impact of gene mutations in the ErbB signalling pathway on overall survival of patients with GBC. The overall survival information was derived from 57 patients with mutations in ErbB pathway genes and 100 patients without such mutations. (D) Impact of mutations in ERBB2 or ERBB3 on overall survival. Data were derived from 21 patients with mutations in ERBB2 or ERBB3 and 136 patients without mutations in either gene.

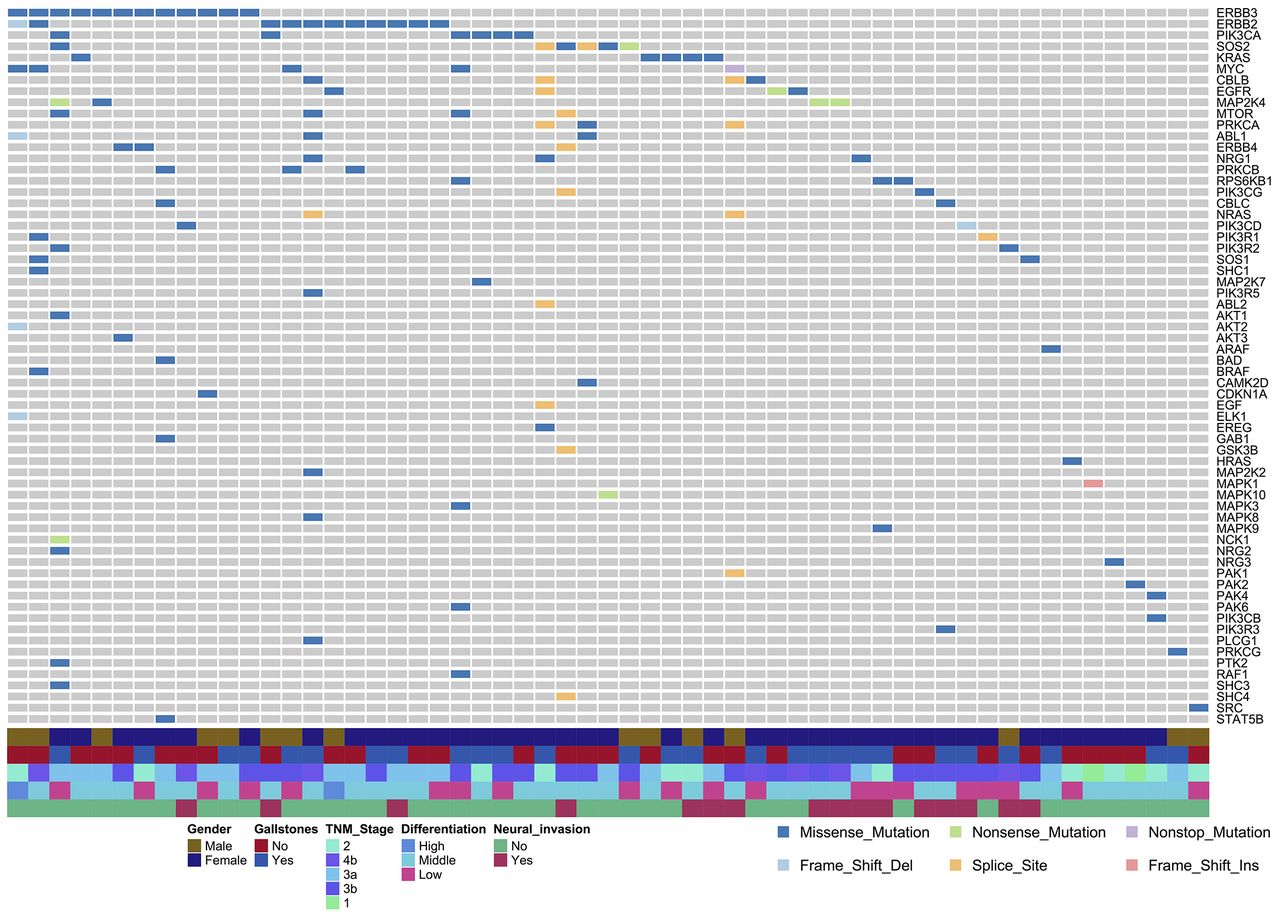
Somatic mutations in the ErbB signalling pathway. A total of 64 genes in the ErbB pathway with at least one non-silent mutation were identified in 157 patients with GBC. Among these patients, 57 (36.3%) had a non-silent mutation. Each column represents one patient; each row represents one gene. Genes are listed based on descending mutation frequency. EV, empty pCMV6 vector; WT, wild-type.
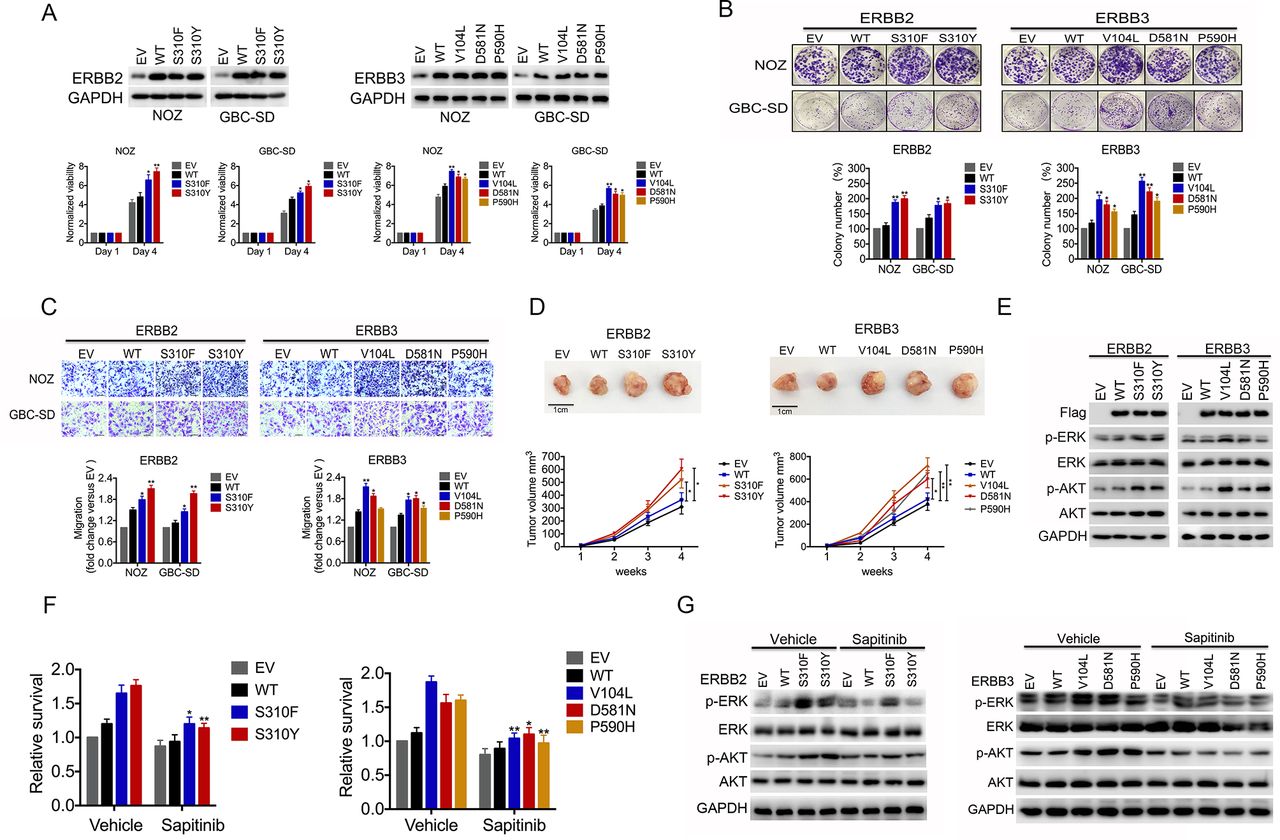
Gallbladder carcinoma (GBC) cells expressing mutant ERBB2/ERBB3 show higher proliferation and sensitivity to anti-ERBB2/ERBB3 inhibitors than wild-type GBC cells. (A) GBC-SD and NOZ cells were transiently transfected with a vector expressing wild-type (WT) or mutant ERBB2 or ERBB3. Expression of relevant proteins was assessed by Western blotting 48 hours after transduction (upper panel), and cell viability was measured using the CCK-8 assay after transduction (lower panel). EV, empty pCMV6 vector. (B) Representative images of colony formation by GSC-SD and NOZ cells expressing WT or mutant ERBB2 or ERBB3 (upper panel). Relative colony counts are presented in the lower panel. (C) Transwell assay to determine the effects of WT or mutant ERBB2 or ERBB3 on the migration of NOZ and GBC-SD cells (upper panel). Relative migration was quantified relative to WT (lower panel). (D) Tumour growth of NOZ cells expressing WT or mutant ERBB2 or ERBB3 in nude mice. Data are presented as the mean±SD from at least five independent samples. *p<0.05, **p<0.01 vs WT. (E) NOZ cells were transfected with plasmids encoding mutant ERBB2 or ERBB3. Then, cell lysates were subjected to immunoblot analysis with antibodies as indicated. (F) NOZ cells expressing mutant ERBB2 or ERBB3 were treated with sapitinib or vehicle for 48 hours. Then, cell viability was assayed using CCK-8. (G) Cell lysates were subjected to immunoblot analysis with antibodies as indicated. Data are represented as the mean±SD from three independent experiments. *p<0.05, **p<0.01 vs WT.
Comparing our data with 30 signatures previously identified revealed four shared signatures: three exhibited cosine similarity of approximately 0.8, and one exhibited cosine similarity of 0.5 (online supplementary figure 3C).12 Signature 2, which was characterised primarily by C>T and C>G mutations at TpCpN trinucleotides, was previously associated with GBC.10 12 This signature has been linked to hyperactivity of APOBEC family cytidine deaminases.13 14
The results of the present study confirm the association between signature 2 and GBC and identify three novel disease-associated mutation signatures. This scenario illustrates the ability of WES to provide reliable, extensive mutation profiling of GBC.
Association of ERBB2/ERBB3 mutations with decreased patient survival
WES revealed that the following genes harboured mutations in greater than 5% of our patients with GBC (online supplementary figure 4): TP53 (27%), KMT2C (11%), SMAD4 (11%), PER3 (8%), ERBB3 (8%), ARID2 (7%), ARID1A (7%) and ERBB2 (7%) (online supplementary tables 7 and 8). All of these genes were identified as potential driver genes by mutsigCV. Hotspot mutations included V104L/M in ERBB3 (5/12) and S310F/Y in ERBB2 (5/11) (figure 1A, B). Genome MUSIC path-scanning15 suggested that these various mutations affect a variety of cancer-related signalling pathways (online supplementary table 9), with the ErbB pathway ranking 21 on this list (False Discovery Rate (FDR), p value=0.0002). Mutations in ERBB3 were identified in 8% of patients; ERBB2, 7%; ERBB1, 2.5%; and ERBB4, 1.9% (online supplementary table 7). Non-silent mutations in the ErbB signalling pathway were identified in 57 of 157 cases, affecting a total of 64 genes (figure 2). These results substantially expand the previously identified set of ErbB-pathway genes associated with GBC.10 Notably, the hotspot mutation S310F/Y was not identified in our previous study.
Supplementary file 6

ERBB2/ERBB3 mutations induce PD-L1 expression and promote immune escape. (A) Microarray expression profiling analysis of NOZ cells stably transfected with the mutant or wild-type ERBB2/ERBB3 gene. (B) NOZ cells were stably transfected with empty vector (EV), wild-type (WT) ERBB2 or ERBB3, mutant ERBB2 (S310F or S310Y) or mutant ERBB3 (V104L, D581N or P590H) and subjected to immunoblotting with anti-PD-L1 antibody. (C, D) NOZ cells were transfected with plasmids encoding mutant or WT ERBB2/ERBB3, and cell surface PD-L1 expression was determined using fluorescence-activated cell sorting. Scale bars indicate median fluorescence intensity. (E) PD-L1 expression was detected by immunohistochemistry in gallbladder carcinoma tissues from patients. Proportions of patients exhibiting low or high PD-L1 expression were compared between those expressing wild-type (WT) or mutant ERBB2/ERBB3. (F) NOZ cells expressing wild-type (WT) or mutant ERBB3 were treated for 72 hours with an Akt or ERK inhibitor. Then, cell lysates were immunoblotted with the indicated antibodies. Data are represented as the mean±SD from three independent experiments. *p<0.05, **p<0.01 vs WT.
Median overall survival time was significantly reduced in patients with ErbB mutations (8.0 months) compared with those without the mutation (12.3 months, log-rank p=0.005, figure 1C). These results are consistent with our previous study.10 Regarding ERBB family genes, low-frequency mutations on ERBB1 or ERBB4 and relative higher-frequency mutations on ERBB2 and ERBB3 were observed. Given that ERBB2 and ERBB3 proteins bind to each other to exert their biological functions,16 we analysed the influence of ERBB2 and ERBB3 mutations on survival and found that ERBB2/ERBB3 mutations were associated with reduced median survival (6.5 vs 11.0 months, log-rank p=0.009; figure 1D and online supplementary table 10). Moreover, the mutation profiles did not correlate with demographics, degree of differentiation or TNM staging (online supplementary table 11). Multivariate Cox proportional hazard modelling confirmed ERBB2/ERBB3 mutations as an independent risk factor of poor patient survival (HR 2.038, 95% CI 1.155 to 3.597; online supplementary table 12) when controlling for several other survival-related factors, including TNM stage, margin status and perineural invasion. In addition, we found that the copy number of ERBB2/ERBB3 by using FISH and qRT-PCR assay was not correlated with the point mutations in ERBB2 and ERBB3 in GBC (online supplementary table 13, supplementary figure 5). This finding is consistent with a previous report that somatic point mutations in the ERBB2/ERBB3 gene often occur in a number of tumour cells without amplification/overexpression of ERBB2/ERBB3.17 18
Supplementary file 7

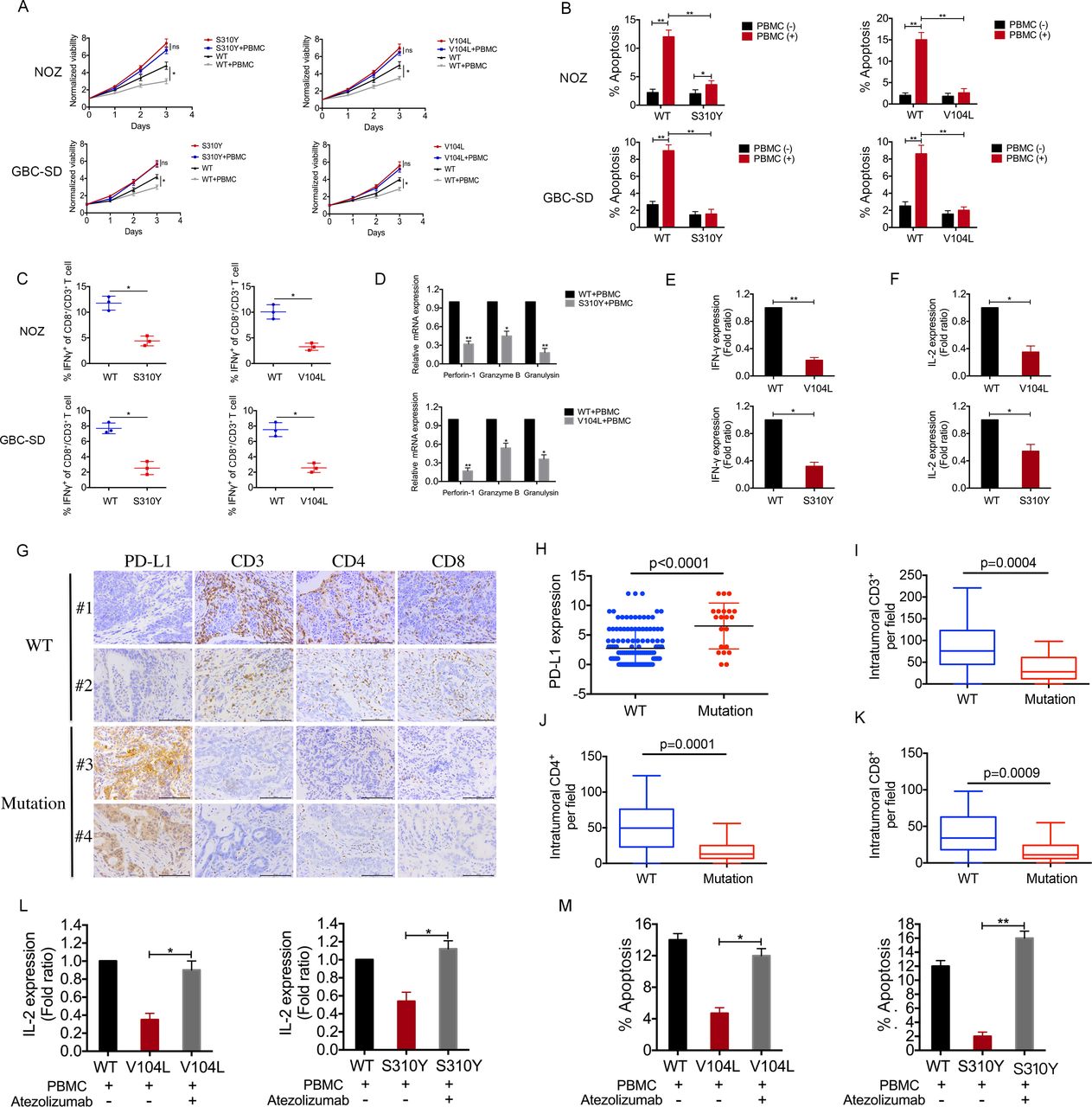
ERBB3/ERBB2 mutations upregulate PD-L1 and suppress anticancer immunity. (A) Gallbladder carcinoma (GBC) cells expressing wild-type (WT) or mutant ERBB3/ERBB3 were co-cultured for 3 days with or without activated peripheral blood mononuclear cells (PBMCs) as described in the Methods section. Carcinoma cell viability was assayed using CCK-8. S310Y, ERBB2 mutant S310Y; V104L, ERBB3 mutant V104L. (B) GBC cells treated as described in panel (A) were collected, stained with FITC–Annexin V and propidium iodide, and analysed for apoptotic state. (C) Quantification of intracellular IFN-γ in CD8+/CD3+ T cells. (D) Quantitative PCR was performed to detect perforin-1, granzyme and granulysin in PBMCs co-cultured with GBC cells. (E, F) Soluble INF-γ and IL-2 levels in the supernatants of co-cultures containing NOZ cells and PBMCs as assayed by ELISA. (G) Sections of formalin-fixed patient tumours carrying wild-type (WT) or mutant ERBB2/ERBB3 were stained against PD-L1, CD3, CD4 or CD8. (H) PD-L1 expression in 157 tumour samples from patients with GBC, of whom 136 expressed wild-type (WT) ERBB2/ERBB3 and 21 expressed mutant ERBB2/ERBB3. (I–K) Quantification of intratumoural CD3+, CD4+ or CD8+ cells per high-power field in the 157 tumour samples. (L) GBC cells expressing wild-type (WT) or mutant ERBB3/ERBB3 were co-cultured with activated PBMCs and then treated for 3 days with the anti-PD-L1 antibody atezolizumab. Soluble IL-2 levels in the supernatant of the co-culture system were assayed by ELISA. (M) Apoptosis of GBC cells in the co-culture system was assayed based on staining with FITC–Annexin V and propidium iodide. Data represent one of three independent experiments in triplicate. **p<0.01 vs WT.
ERBB2/ERBB3 mutations promote GBC cellular proliferation and enhance sensitivity to anti-ERBB2/ERBB3 inhibitors in vitro
We expressed the S310Y and S310F mutants of ERBB2 and the V104L, D581N and P590H mutants of ERBB3 in GBC-SD and NOZ cells, which express wild-type and low levels of endogenous ERBB2 and ERBB3 (online supplementary figure 10A). Ectopic expression of these mutants, especially the two hotspot mutants S310Y and V104L, increased cell proliferation, colony formation and migration (figure 3A–C). NOZ cells xenografted onto nude mice grew significantly faster if they expressed these mutant proteins rather than the wild-type protein (figure 3D). The MAPK and PI3-K/Akt pathways are major signalling pathways downstream of ERBB2/ERBB3 activation, and both increase cell proliferation and inhibit cellular apoptosis.19 Expression of mutant ERBB2 or ERBB3 was associated with increased p-AKT and p-ERK levels based on Western blotting (figure 3E, online supplementary figure 6A).
Supplementary file 8
Supplementary file 9

![[gutjnl-2018-316039supp003.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
![[gutjnl-2018-316039supp004.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
![[gutjnl-2018-316039supp005.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
![[gutjnl-2018-316039supp006.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC6/embed/inline-supplementary-material-6.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316039supp007.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC7/embed/inline-supplementary-material-7.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316039supp008.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC8/embed/inline-supplementary-material-8.jpg?download=true){kind=link}
![[gutjnl-2018-316039supp009.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC9/embed/inline-supplementary-material-9.jpg?download=true){kind=link}
{kind=link}
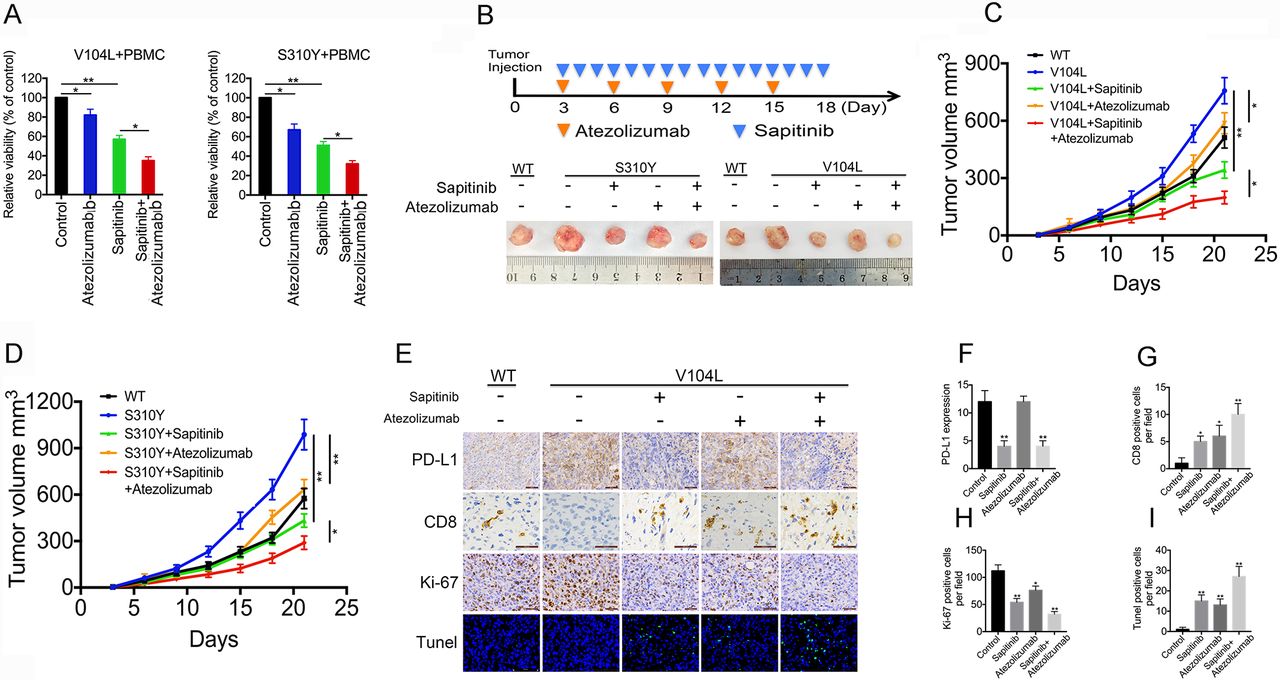
PD-L1 blockade potentiates ERBB2/ERBB3-targeted therapy in gallbladder cells expressing mutant ERBB2/ERBB3. (A) Relative viability of NOZ cells expressing mutant ERBB2/ERBB3 and co-cultured for 72 hours with peripheral blood mononuclear cells (PBMCs) in the presence of atezolizumab, sapitinib or both. (B) Representative images of gallbladder carcinoma xenografts in CD34+ humanised NCG mice at the indicated times of sapitinib and/or atezolizumab treatment (n=5). (C, D) Volume of xenografts in mice treated with sapitinib and/or atezolizumab. (E) Immunohistochemistry of xenografts to visualise CD8, PD-L1 and Ki-67 as well as TUNEL-positive cells. (F) PD-L1 expression in xenografts after sapitinib and/or atezolizumab treatment. (G–I) Quantitation of CD8-positive, Ki-67-positive or TUNEL-positive cells in xenografts after sapitinib and/or atezolizumab treatment. Data represent one of three independent experiments in triplicate. *p<0.05, **p<0.01. WT, wild-type.
Activating ERBB2 mutations are highly sensitive to pan-ERBB inhibitors in lung cancer, breast cancer and colorectal cancer, and ERBB2 inhibitors are being assessed in clinical trials for various cancer types.20–22 Therefore, we tested the sensitivity of GBC cells expressing mutant ERBB2/ERBB3 to the ERBB2/ERBB3 tyrosine kinase inhibitor sapitinib.23 24 Sapitinib significantly blocked tumour growth and inhibited ERK and Akt phosphorylation levels in GBC cells with ERBB2/ERBB3 mutations; however, cell proliferation and p-ERK or p-Akt levels in cells expressing wild-type ERBB2/ERBB3 were not affected (figure 3F, G, online supplementary figures 6B, 7A,B).
Induction of PD-L1 expression by ERBB2/ERBB3 mutants
To investigate which genes may be differentially regulated in the presence of ERBB2/ERBB3 mutations, we compared the microarray profiles of GBC cell lines expressing wild-type versus mutant ERBB2/ERBB3 (figure 4A, online supplementary tables 14, 15). Functional analysis of gene ontology biological processes showed that the differentially expressed genes found in mutant ERBB2/ERBB3 cells were closely related to immune response (online supplementary figure 8). In particular, we found that PD-L1, which plays crucial roles in the immune response, was among the upregulated genes (online supplementary figure 9A). Up-regulated PD-L1 expression was further confirmed by qRT-PCR, western blotting and fluorescence-activated cell sorting (figure 4B–D, online supplementary figure 9B–D). Immunohistochemistry also revealed high PD-L1 expression in 14 of 21 (66.7%) GBC tumours expressing mutant ERBB2/ERBB3, which was greater than the 30 of 136 (22.1%) samples expressing wild-type ERBB2/ERBB3 (p<0.01, figure 4E). In GBC cell lines, PD-L1 localised to the cell membrane and cytoplasm, and its expression correlated with ERBB2 and ERBB3 expression (Spearman correlation coefficient R=0.9; p<0.05; online supplementary figure 10A–C). Knockdown of ERBB2/ERBB3 or treatment with sapitinib significantly decreased PD-L1 expression in NOZ cell lines (online supplementary figure 10D–H). The Akt inhibitor MK-2206 suppressed expression of both p-AKT and PD-L1, whereas the ERK inhibitor SCH772984 reduced levels of p-ERK without affecting PD-L1 levels (figure 4F, online supplementary figure 9E). These results indicate that activating ERBB2/ERBB3 mutations may induce PD-L1 expression mainly through the PI3K/Akt signalling pathway.
Supplementary file 11
![[gutjnl-2018-316039supp010.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC10/embed/inline-supplementary-material-10.jpg?download=true){kind=link}
Supplementary file 12
![[gutjnl-2018-316039supp011.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC11/embed/inline-supplementary-material-11.jpg?download=true){kind=link}
Upregulation of PD-L1 associated with ERBB3/ERBB2 mutations suppresses anticancer immunity
We performed a T-cell-mediated killing assay to determine whether the upregulation of PD-L1 associated with ERBB2/ERBB3 mutations affects T-cell function. In a co-culture system, PBMCs inhibited proliferation and promoted apoptosis of GBC cells expressing wild-type ERBB2/ERBB3 but not in tumour cells expressing mutant ERBB2/ERBB3 (figure 5A,B). In addition, the cytotoxic T-cell activity of PBMCs was reduced in co-cultures of GBC cells expressing mutant ERBB2/ERBB3 compared with co-cultures with cancer cells expressing wild-type genes (figure 5C–F), which was also observed in GBC samples (figure 5G).
PD-L1 expression was significantly higher in patient tumour samples expressing mutant ERBB2/ERBB3 than in those expressing the wild-type genes (figure 5H). Conversely, levels of CD3, CD4 and CD8 were significantly reduced in tumour samples expressing the mutant genes (figure 5I–K, online supplementary table 16). Treating PBMC/GBC co-cultures with the anti-PD-L1 antibody atezolizumab significantly enhanced the release of IL-2 into the supernatant and promoted T-cell-mediated killing (figure 5L,M). These results suggest that ERBB2/ERBB3 mutations are associated with activation of the PD-1/PD-L1 axis, which suppresses T-cell function and helps GBC cells escape antitumour immune responses.
PD-L1 blockade potentiates anti-ERBB2/ERBB3 therapy in the presence of ERBB2/ERBB3 mutations
The above findings led us to hypothesise that PD-L1 blockade can enhance the efficacy of anti-ERBB therapy in GBC cells carrying mutant ERBB2/ERBB3. Indeed, exposing NOZ cells that express mutant ERBB2/ERBB3 in co-culture with PBMCs to atezolizumab decreased their cellular viability by 30%. Exposing these cells to sapitinib decreased their cellular viability by 45% (figure 6A), and exposure to both inhibitors decreased cellular viability by 70%. Either inhibitor alone significantly inhibited the growth of tumours expressing mutant ERBB2 or ERBB3 in CD34+ humanised NCG mice with an intact immune system, and combination therapy exhibited synergistic inhibition of tumour growth (figure 6B–D). Immunohistochemistry revealed that tumour treatment with both inhibitors in combination led to significantly fewer Ki-67-positive cells and more CD8-positive and TUNEL-positive cells than treatment with a single inhibitor (figure 6E–I). None of the treatments adversely affected weight or liver or kidney function in the tumour-bearing mice (online supplementary figure 11). These results suggest that anti-PD-L1 agents can safely and effectively potentiate GBC therapies targeting ERBB2/ERBB3 in the presence of mutant ERBB2/ERBB3.
Supplementary file 13
![[gutjnl-2018-316039supp012.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC12/embed/inline-supplementary-material-12.jpg?download=true){kind=link}
Discussion
Genomic alterations in GBC appear to be the core elements that commit tumour cells to malignant transformation.10 To overcome the limitations of small samples and lack of detailed information between genotype and clinical phenotype in previous studies, we used WES to screen for gene variants in a larger pool of 157 patients with GBC. We found that the ErbB signalling pathway is an essential pathway associated with GBC malignancy, and mutations in this pathway are correlated with poor survival. In this large data set, we also confirmed the high rate of C>T and G>A transitions in GBC that we observed in our previous, smaller study.10 Moreover, we discovered evidence that ERBB2/ERBB3 mutations may influence the ability of tumours to evade immune responses, which may point to novel targets for cancer therapy.
Here, we identified four mutation signatures. Signature 2 was previously reported to be associated with GBC,10 whereas signatures 1, 4 and 17 are novel. Signature 1 is characterised by the prominence of C>T substitutions at NpCpG trinucleotides and is likely related to the relatively elevated rate of spontaneous deamination of 5-methyl-cytosine.12 The investigations of signatures 4 and 17 were limited. Our analysis also revealed that the following genes were mutated at a high frequency in our large cohort of patients: TP53 (27%), KMT2C (11%), SMAD4 (11%), PER3 (8%), ARID2 (7%), ARID1A (7%) and genes of the ErbB signalling pathway (36.3%). Multivariate Cox regression identified ERBB2 or ERBB3 mutations as independent risk factors of poor patient survival.
ERBB2 and ERBB3 mutations have been implicated in a variety of cancers.16 25 26 For example, the somatic mutation V104L in ERBB3 may induce malignant transformation in a ligand-independent fashion, and ERBB3 small-molecule inhibitors are effective in colon and breast cancers harbouring the mutation.16 The S310F/Y mutation in ERBB2 contributes to breast and lung cancer by altering phosphorylation of the protein’s C-terminal tail, and the oncogenic effects of these mutations can be reduced by small-molecule inhibitors.26 27 The present study together with our previous work10 identified S310F/Y mutations in ERBB2 and V104L/M mutations in ERBB3 as hotspot mutations in GBC. Our experiments in gallbladder cell cultures expressing ERBB2 S310Y/F and ERBB3 V104L revealed that these gene mutations are associated with enhanced proliferation and migration abilities and that these effects involve activation of PI3K/Akt and ERK signalling pathways. These findings indicate an important role of ERBB2/ERBB3 mutations in GBC pathogenesis and encourage future work to explore ERBB2/ERBB3 as a treatment target. We are currently conducting a phase I study to evaluate the potential efficacy of the pan-ERBB receptor inhibitor KBP-5209 in patients with GBC who carry ERBB2/ERBB3 mutations (ClinicalTrials.gov Identifier: NCT 02442414).
Tumour cells have the ability to induce multiple immune inhibitory checkpoints to establish an immunosuppressive microenvironment.28 PD-L1 is a T-cell inhibitory ligand that interacts with its receptor PD-1 to maintain self-tolerance and protect against excessive tissue damage induced by immune responses. Thus, PD-L1 functions as an immune checkpoint under physiological conditions.29 The expression of this checkpoint regulator is altered by oncogenic mutations in numerous cancers. For example, in non-small cell lung carcinoma, EGFR and KRAS mutations are associated with increased PD-L1 expression levels.1 In lung adenocarcinoma, KRAS and TP53 mutations are associated with high PD-L1 levels.30 Consistent with these data, we showed increased PD-L1 expression in the primary GBC lesions carrying ERBB2/ERBB3 mutations. These findings in numerous cancers highlight the potential efficiency of targeting PD-L1 as an antitumour therapy. In clinical trials, antibody blockade of PD-L1 was shown to improve therapeutic outcomes in patients with metastatic melanoma, renal cell carcinoma, non-small cell lung cancer and other types of cancer.31–34
Effectively exploiting PD-L1 as a drug target requires detailed understanding of how it participates in oncogenesis and progression. In several types of haematological neoplasms and other solid tumours,35–38 PD-L1 is regulated by the MEK–ERK and PI3K/Akt signalling pathways. In the present study, we found that ERBB-induced PD-L1 expression may depend on PI3K/Akt rather than the MEK-ERK signalling pathway. This finding suggests that PD-L1 expression and activity are varied across different cancers, which should be explored in future studies to ensure effective and specific therapeutic effects for patients with a given cancer type. In the present study, we first identified that ERBB2/ERBB3 mutations promote immune evasion, which may explain how they contribute to GBC development. We found that ERBB2/ERBB3 mutations inhibited the ability of tumour-reactive T cells to target tumour cells for apoptosis through upregulating PD-L1 and attenuated the release of IFN-γ and IL-2 into co-culture supernatants and in tumour cells. The results in the present work reveal a model in which ERBB2/ERBB3 mutations induce PD-L1 expression, which subsequently triggers CD8+ T-cell apoptosis and reduces the production of INF-γ and IL-2.
Consistent with this model, we found that sapitinib and atezolizumab, which reduced PD-L1 expression, restored the antitumour activities of T cells in our GBC co-culture system and in xenografts in CD34+ HuNCG mice in the presence of ERBB2/ERBB3 mutations. Treating the carcinoma cells expressing mutant ERBB2/ERBB3 led to tumours that were smaller than control tumours expressing wild-type ERBB2/ERBB3. Of note, PD-L1 blockade potentiated ERBB2/ERBB3-targeted therapy of ERBB2/ERBB3-mutant GBC cells. These results suggest that ERBB and/or PD-L1 at least partially activate and contribute to the development of GBC in the absence of ERBB2/ERBB3 mutations. Further work is warranted to understand how these two inhibitors promote apoptosis in GBC. Regardless of the mechanisms, our results justify further work exploring potential therapies that target ERBB and/or PD-L1 in GBC.
The current study identified high-frequency ERBB2/ERBB3 mutations in patients with GBC, and these mutations represent independent risk factors of overall survival for patients. ERBB2/ERBB3-activating mutations promote tumour cell growth and migration as well as upregulate PD-L1 expression to induce immune evasion of GBC cells. These findings suggest that ERBB2/ERBB3 mutations may be biomarkers for identifying patients sensitive to therapies targeting ERBB2/ERBB3 and PD-L1.
Supplementary file 10
![[gutjnl-2018-316039supp013.jpg]](https://gut.bmj.com/content/gutjnl/68/6/1024/DC13/embed/inline-supplementary-material-13.jpg?download=true){kind=link}
References
Footnotes
ML, FL, FZ, WZ and XJ contributed equally.
Contributors YBL, YL and HL conceived of the study. WZ, XJ, JZ, CL, HL, YL and YBL directed the study. ML, FL, FZ, HL, YL and YBL contributed to the project design. ML, FL, FZ, YY, KQ, YW, QM, TW and LB performed experiments. ML, FL, ZW and XS performed the bioinformatics data analysis. YZ, RY, YG, YL, YJ, HL, SX, YY, YZ, LJ, YuH and YaH contributed samples, data and comments on the manuscript. ML, FL, FZ, WL, SC, JG, JZ and WG analysed and interpreted data. XW, XS, YZ, RY and YG contributed reagents/materials/analysis tools. ML, FL and FZ wrote the manuscript.
Funding This study was supported by the National Natural Science Foundation of China (Nos. 81773043, 31620103910, 91440203, 81672404, 81502432 and 81502433), the Shanghai Key Laboratory of Biliary Tract Disease Research Foundation (No. 17DZ2260200), the Shanghai Sailing Program (No. 18YF1415100), the Shanghai Jiao Tong University Medical-engineering Cross Program (YG2014MS69), the development fund for Shanghai talents (No.201608) and the Shanghai Rising-Star Program (No. 15QA1403100).
Competing interests None declared.
Ethics approval The study was approved by the ethics committees of the Xinhua Hospital, Zhongshan Hospital and Eastern Hepatobiliary Surgery Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.