Article Text

Abstract
Background and aims The crosstalk between cancer stem cells (CSCs) and their niche is required for the maintenance of stem cell-like phenotypes of CSCs. Here, we identified E26 transformation-specific homologous factor (EHF) as a key molecule in decreasing the sensitivity of pancreatic cancer (PC) cells to CSCs’ niche stimulus. We also explored a therapeutic strategy to restore the expression of EHF.
Design We used a LSL-KrasG12D/+mice, LSL-Trp53R172H/+ and Pdx1-Cre (KPC) mouse model and samples from patients with PC. Immunostaining, flow cytometry, sphere formation assays, anchorage-independent growth assay, in vivo tumourigenicity, reverse transcription PCR, chromatin immunoprecipitation (ChIP) and luciferase analyses were conducted in this study.
Results CXCL12 derived from pancreatic stellate cells (PSCs) mediates the crosstalk between PC cells and PSCs to promote PC stemness. Tumorous EHF suppressed CSC stemness by decreasing the sensitivity of PC to CXCL12 stimulus and inhibiting the crosstalk between PC and CSC-supportive niches. Mechanically, EHF suppressed the transcription of the CXCL12 receptor CXCR4. EHF had a cell autonomous role in suppressing cancer stemness by inhibiting the transcription of Sox9, Sox2, Oct4 and Nanog. Rosiglitazone suppressed PC stemness and inhibited the crosstalk between PC and PSCs by upregulating EHF. Preclinical KPC mouse cohorts demonstrated that rosiglitazone sensitised PDAC to gemcitabine therapy.
Conclusions EHF decreased the sensitivity of PC to the stimulus from PSC-derived CSC-supportive niche by negatively regulating tumorous CXCR4. Rosiglitazone could be used to target PC stem cells and the crosstalk between CSCs and their niche by upregulating EHF.
- pancreatic cancer
- stem cells
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Pancreatic cancer (PC) is one of the leading causes of cancer-related death and is projected to become the second most lethal tumour by the year 2030.
Cancer stem cells (CSCs) contributed to PC recurrence and metastasis.
PC stemness is regulated by the aberrant activation of cell-intrinsic signal pathways and the crosstalk between CSCs and their niche.
Patients with PC and low tumorous E26 transformation-specific homologous factor (EHF) expression gained poor overall and relapse-free survival.
What are the new findings?
Tumorous EHF decreased the sensitivity of PC to pancreatic stellate cell (PSC)-derived CXCL12 stimulus, which suppressed cancer stemness by inhibiting the crosstalk between PC and CSC-supportive niches.
EHF transcriptionally suppressed CXCR4, which is the receptor of CXCL12.
EHF also suppressed cancer stemness in a cell autonomous manner by transcriptionally suppressing Sox9, Sox2, Oct4 and Nanog.
Rosiglitazone suppressed PC stemness and inhibited the crosstalk between PC and PSCs by upregulating EHF.
How might it impact on clinical practice in the foreseeable future?
Our study identified that EHF suppressed cancer stemness from intrinsic and extrinsic pathways, which could be a promising target in PC therapy.
Rosiglitazone could be used as a new therapeutic method in clinical practice to target pancreatic cancer stem cells and the crosstalk between CSCs and their niche.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal tumour with aggressive clinical courses, poor prognosis and limited treatment options. Chemotherapy resistance and tumour relapse are still two unresolved problems in PDAC treatment.1 2 Cancer stem cells (CSCs) contribute to PDAC recurrence and metastasis and cause resistance to chemotherapy.3–6
CSCs are regulated by the aberrant activation of cell-intrinsic signal pathways, including NOTCH, WNT and STAT3 pathways, and the overexpression of OCT4, SOX2, NANOG, KLF4, c-MYC among others.3 7 The recent insights into the complex nature of cancer stemness reveal that CSC phenotype is also regulated by cell-extrinsic factors derived from stromal cells.8–11 The major cell types of PDAC stroma are pancreatic stellate cells (PSCs).9 12 13 PSCs can secrete prostemness cytokines, such as interleukin (IL)-6, IL-8, tumour growth factor beta 1 (TGF-β1) and CXCL12, which form the CSC niche and participate in the active crosstalk with cancer cells within the tumour microenvironment.14–16 The majority of anti-CSC therapeutic strategies focus on targeting cell-intrinsic stemness-associated genes. However, most of these genes are shared between CSCs and normal stem cells. Therefore, the side effect of anti-CSC therapy remains a major concern that restricts its clinical application.17 Targeting the crosstalk between pancreatic cancer (PC) and its stemness-supporting niche may provide new therapeutic strategies for the prevention of PC progression.
Epithelium-specific E26 transformation-specific (ETS) factor family member 3 or ESE3/E26 transformation-specific homologous factor (EHF) is a member of the ETS gene superfamily.18 Our previous work has demonstrated EHF as a tumour suppressor in PDAC. In PDAC, EHF promotes E-cadherin expression and suppresses epithelial–mesenchymal transition.19 Furthermore, EHF deficiency induces the conversion and expansion of regulatory T cells (Treg) and myeloid-derived suppressor cells (MDSCs) by inhibiting TGF-β1 and granulocyte-macrophage colony stimulating factor (GM-CSF) secretion.20 In prostate cancer, EHF plays a vital role in the inhibition of cell-intrinsic CSC signal by suppressing STAT3 and repressing the expression of TWIST1, ZEB2, BMI1 and POU5F1.21–23 However, the role of EHF in pancreatic CSC regulation is not fully understood. Although the critical function of EHF has been verified in different tumour types,19–25 no clinical and translational research involving EHF as a therapeutic target has been conducted.
In this study, we demonstrated that EHF could play a cell autonomous function and inhibit PDAC stemness by disrupting the crosstalk between CSCs and their PSC niche. Tumorous EHF decreased the sensitivity of PDAC to PSC-derived CXCL12 by repressing the CXCR4 expression. Moreover, we identified peroxisome proliferator-activated receptor gamma (PPAR-γ) ligand rosiglitazone as a promising suppressor of PDAC stemness by upregulating EHF.
Results
Tumorous EHF is negatively correlated with stemness profiles in PDAC tissue
An immunohistochemical multiplex assay was conducted in archived tissues from a retrospective cohort of 93 patients with PDAC to examine the correlation between the expression of tumorous EHF and the proportion of pancreatic cancer stem cells (PCSCs). The frequencies of CD133+ and aldehyde dehydrogenase 1(ALDH1+)cells in the high-EHF group were significantly decreased compared with those in the low-EHF group (both p<0.0001; figure 1A,B and online supplemental figure S1A). Furthermore, fresh PDAC tissues from a prospective cohort of 39 patients were collected and analysed (figure 1C). As shown in figure 1D–F and online supplemental figure S1B, tumorous EHF IHC score was inversely correlated with the proportion of tumorous CD133+, ALDH+ and ESA+CD44+CD24+ cells in the prospective cohort. These results confirmed the findings from the archived PDAC tissues. Therefore, our results suggested that tumorous EHF negatively correlated with stemness profiles in PDAC tissues. Besides, the clinical significance of EHF expression and PCSCs is shown in figure 1G,H, online supplemental figure S1C–F and online supplemental tables S1 and S2).
Supplemental material
Supplemental material

Tumorous EHF is negatively correlated with stemness profiles in PDAC tissue. (A,B) Multiplex fluorescent IHC staining (left) of EHF expression and the accumulation of CD133+ cells (A) and ALDH1+ cells (B) in tumour tissues. The representative images from 93 pancreatic cancer cases are shown. The arrowheads indicate CD133+ cells and ALDH1+ cells. Bars, 200 µM. Non-paired Student’s t-test was used as statistical analysis; n=93, ****p<0.0001. (C–F) Single-cell suspensions were prepared from 39 cases of fresh PDAC tissues and stained with ALDEFLUOR or specific antibodies against three CSC subsets (ALDH+ cells, CD133+ cells and CD44+ CD24+ cells). Representative IHC staining of EHF is shown (C). Bars, 200 µm. Representative histogram and dot plots of CD133+ cells (D, left), ALDH+ cells (E, left) and CD44+CD24+ cells (gated on ESA+ epithelial cells; F, left). Spearman correlation analysis between EHF IHC score and the proportions of CD133+ cells (D, right), ALDH+ cells (E, right) and ESA+CD44+CD24+ cells (F, right); n=39. Kaplan-Meier OS (G) and RFS (H) for different levels of EHF based on the log-rank statistic test (p<0.05). Patients were divided into EHF-low and EHF-high groups based on the multiplex fluorescent IHC results. ALDH, aldehyde dehydrogenase; CSC, cancer stem cell; DAPI, 4’,6-diamidino-2-phenylindole; EHF, E26 transformation-specific homologous factor;IHC, immunohistochemistry; OS, overall survival;PDAC, pancreatic ductal adenocarcinoma; RFS, relapse free survival; SSC, side scatter.
Tumorous EHF negatively regulates PC stemness
PDAC-EHF/short hairpin EHF (shEHF) cell lines were established (online supplemental figure S2A) to determine whether tumorous EHF regulated PDAC stemness. The percentage of CD44+CD24+ cells in PDAC-EHF significantly decreased compared with that in the PDAC-vector control group (figure 2A). By contrast, the percentage of CD44+CD24+ cells in PDAC-shEHF significantly increased compared with that of PDAC-scramble (figure 2B). Similarly, EHF negatively regulated the ALDH activity (figure 2C,D) and the proportions of CD133+ cells (figure 2E,F). In vitro sphere formation assay demonstrated that EHF negatively regulated the cellular sphere formation capacity of PDAC (figure 2G,H). In in vivo limited dilution assay, the ectopic expression of EHF significantly reduced, whereas the knockdown of EHF increased the tumour incidence (figure 2I). This result suggested that EHF suppressed CSC stemness in PDAC. Quantitative PCR (Q-PCR) and western blot demonstrated that EHF negatively regulated stemness-related genes (Sox9, Sox2, Oct4 and Nanog) (figure 2J,K) while increasing the expression of differentiation markers (online supplemental figure S2B). These findings were further confirmed in the other PC cell lines and two PDX cell lines (online supplemental figures S3 and S4). Chromatin immunoprecipitation (ChIP) analyses revealed that EHF directly bound to the promoter region of Sox9, Sox2, Oct4 and Nanog (figure 2L).
Supplemental material
Supplemental material
Supplemental material

Tumorous EHF negatively regulates pancreatic cancer stemness. (A,B) The proportion of CD44+CD24+ cells in PANC-1-vector/EHF, MiaPaca-2-vector/EHF, BxPC-3-scramble/shEHF and SW1990-scramble/shEHF cells were analysed using flow cytometry. Representative dot plots (A, left; B, left) and percentage of CD44+CD24+ cells (A, right; B, right) are shown. (C,D) The proportion of ALDH+ cells in indicated cells were analysed using flow cytometry. Representative dot plots (C, left; D, left) and percentage of ALDH+ cells (C, right; D, right) are shown. (E,F) The proportion of CD133+ cells in indicated cells were analysed using flow cytometry. Representative histograms (E, left; F, left) and percentage of CD133+ cells (E, right; F, right) are shown. (G,H) Sphere formation assays were performed in indicated cell lines. Representative images (G, left; H, left) and sphere number analysis (G, right; H, right) are shown. Bars:100 µm. (I) In vivo limited dilution assays were performed to determine the effects of EHF overexpression or EHF depletion on CSC self-renewal of PANC-1 cells. Representative tumour incidence and CSC probabilities are shown. (J) Q-PCR on EHF and the stemness markers of Sox9, Sox2, Nanog and Oct4 were performed in indicated cells. Actin was used as internal control. (K) Western blot on EHF, Sox9, Sox2, Nanog and Oct4 were analysed in indicated cell lines. β-Tubulin was used as loading control. representative results are shown. (L) ChIP assay was performed to validate transcriptional regulation on Sox9, Sox2, Nanog and Oct4 by EHF. Predicted EBSs in the promoters of human Sox9, Sox2, Nanog and Oct4 (L, left). Binding of EHF to the promoters of the indicated genes in PANC-1 cells determined by ChIP (L, medium). IgG was used as negative control and anti-RNA polymerase Ⅱ was used as PC. AcH3 and H3K27me3 occupancy on the identified EBSs in the promoters of the indicated genes in PANC-1 cells determined by ChIP (L, right). Representative results are shown. All experiments were repeated three times independently. Paired Student’s t-test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ALDH, aldehyde dehydrogenase; ChIP, chromatin immunoprecipitation; CSC, cancer stem cell;EBS, EHF-binding site; EHF, E26 transformation-specific homologous factor; PC, positive control;Q-PCR, quantitative PCR; short hairpin EHF, shEHF; SSC, side scatter; TSS, transcriptional start site.
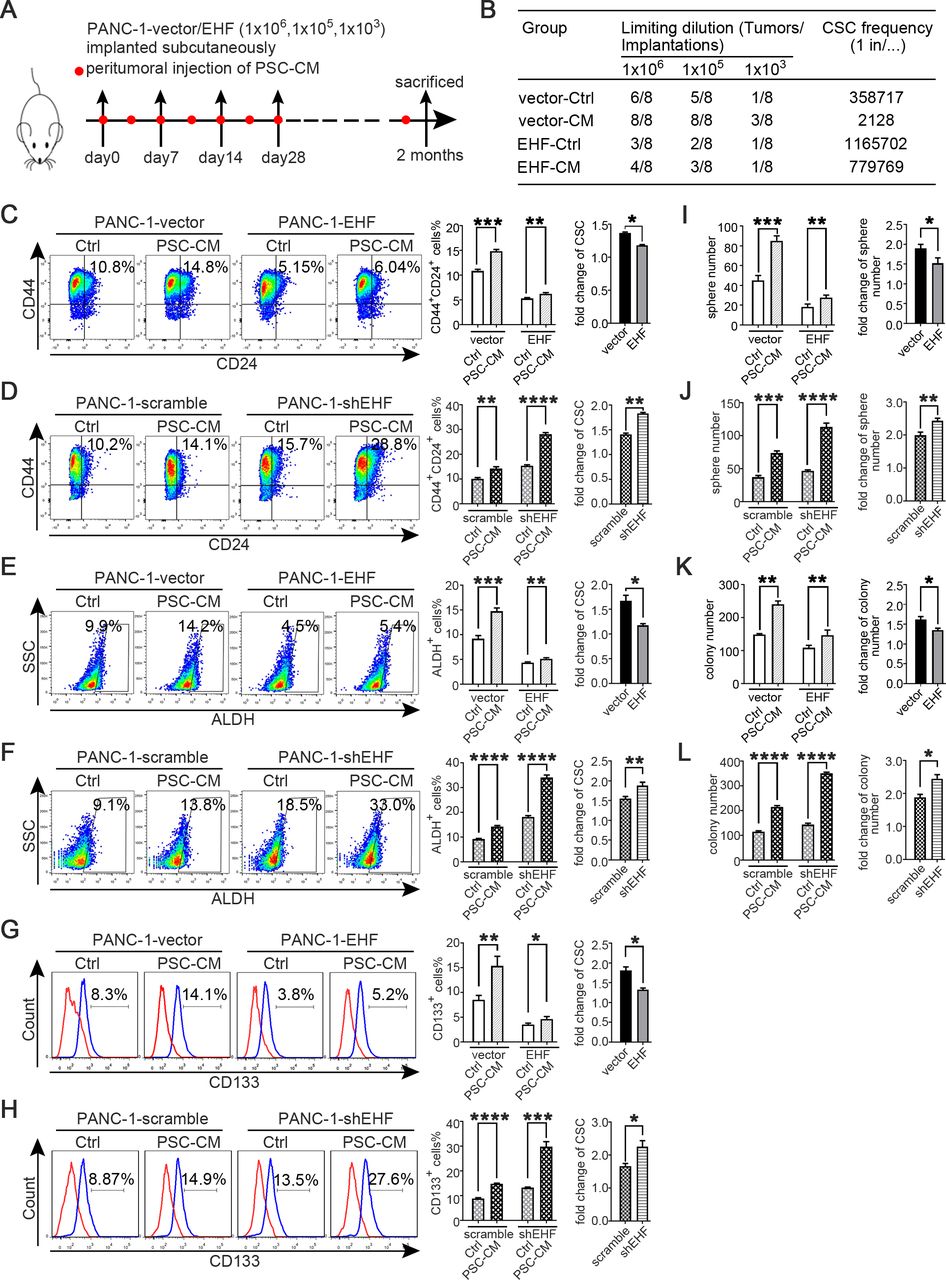
Tumorous EHF suppresses the crosstalk between PDAC and PSCs. (A,B) In vivo limited dilution assay was performed to determine the effects of PSC-CM on CSC self-renewal of PANC-1-vector/EHF. Representative tumour incidence and CSCs probabilities are shown. All experiments were repeated three times independently. (C–H) PANC-1-vector, PANC-1-EHF, PANC-1-scramble and PANC-1-shEHF were cultured with PSC-CM or the Ctrl medium. The percentages of PCSCs in each cell line under each treatment are shown; the fold change of the percentage of PCSCs in each cell line after culturing with PSC-CM was calculated: (C,D) CD24+CD44+ cells, (E,F) ALDH+ cells and (G,H) CD133+ cells. Representative dot plots/histogram (left), the statistical analysis of CSC percentage of each group (medium) and the statistical analysis of the fold change in each cell line (right). (I,J) Statistical analysis of the sphere number of each cell line under the treatment of serum-free medium and serum-free medium with PSC-CM added (left), statistical analysis of the fold change of sphere number after culturing with serum-free medium containing PSC-CM in each cell line(right). (K,L) Statistical analysis of the soft agar colony number of each cell line under the treatment of Ctrl medium and PSC-CM (left). Statistical analysis of the fold change of colony number after culturing with PSC-CM in each cell line (right). Paired Student’s t-test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ALDH, aldehyde dehydrogenase; CM, conditioned medium; CSC, cancer stem cells; Ctrl, control; EHF, E26 transformation-specific homologous factor; PCSCs, pancreatic cancer stem cells; PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cell; shEHF, short hairpin EHF; SSC, side scatter.
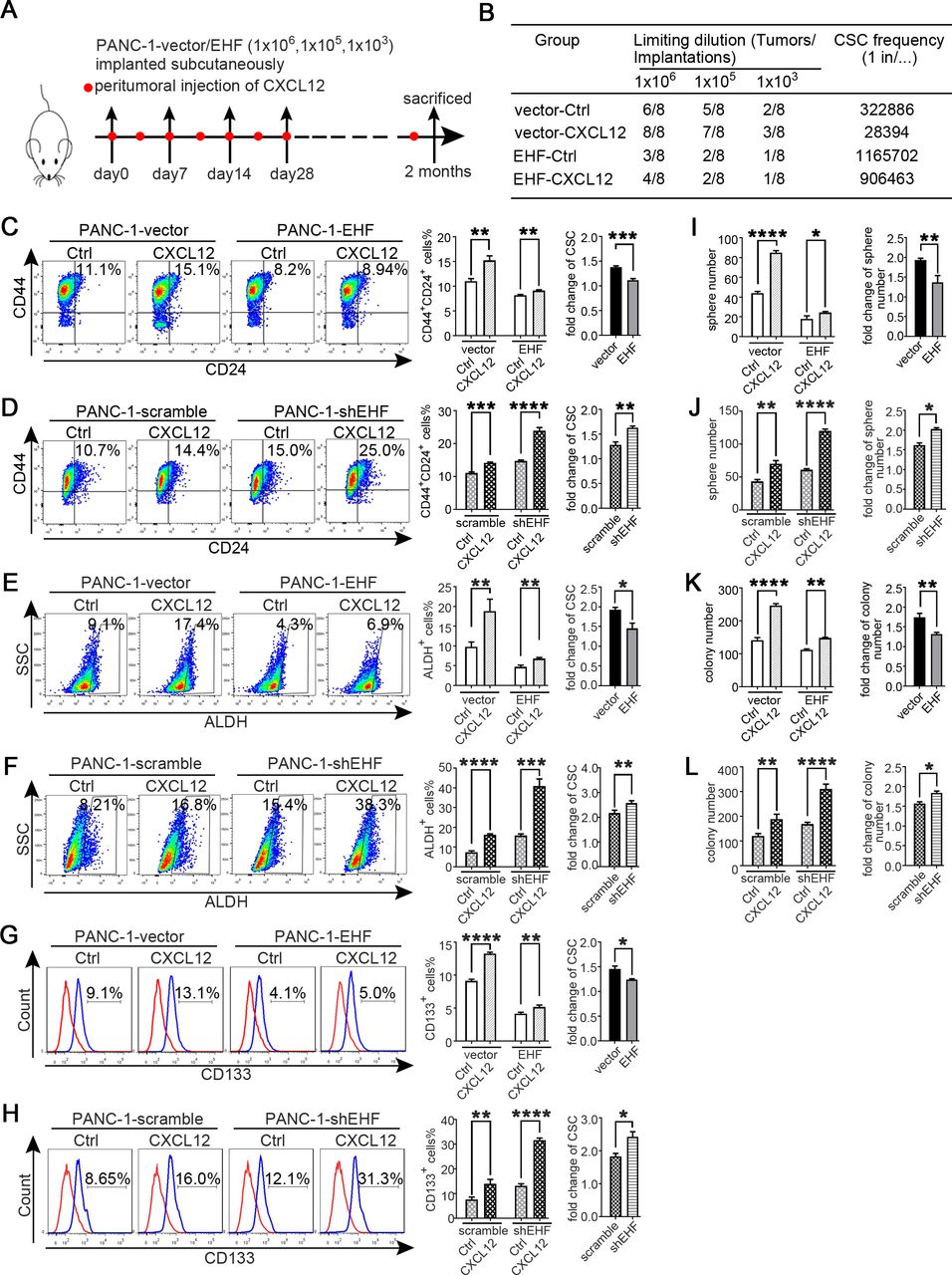
Tumorous EHF abrogates the sensitivity of PDAC to PSC-derived CXCL12 stimulus. (A,B) In vivo limited dilution assay was performed to determine the effects of human recombinant CXCL12 on CSC self-renewal of PANC-1-vector/EHF. Ctrl medium was used as the control of CXCL12.Tumour incidence and CSC probabilities were shown. (C–H) PANC-1-vector, PANC-1-EHF, PANC-1-scramble and PANC-1-shEHF were cultured with medium containing CXCL12 or the Ctrl medium. The percentages of PCSCs in each cell line under each treatment are shown; the fold change of the percentage of PCSCs in each cell line after culturing with medium containing CXCL12 was calculated: (C,D) CD24+CD44+ cells, (E,F) ALDH+ cells and (G,H) CD133+ cells. Representative dot plots/histogram (left), the statistical analysis of CSC percentage of each group (medium) and the statistical analysis of the fold change in each cell line (right). (I,J)Statistical analysis of the sphere number of each cell line under the treatment of serum-free medium and serum-free medium with CXCL12 added (left), statistical analysis of the fold change of sphere number after culturing with serum-free medium containing CXCL12 in each cell line (right). (K,L) Statistical analysis of the soft agar colony number of each cell line under the treatment of Ctrl medium and medium containing CXCL12 (left), statistical analysis of the fold change of colony number after culturing with medium containing CXCL12 in each cell line (right). All experiments were repeated three times independently. Paired Student’s t-test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ALDH, aldehyde dehydrogenase; CSC, cancer stem cell; Ctrl, control; EHF, E26 transformation-specific homologous factor; PCSCs, pancreatic cancer stem cells; PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cell; shEHF, short hairpin EHF; SSC, side scatter.
Tumorous EHF suppresses the crosstalk between PDAC and PSCs
PSCs can support PDAC stemness through paracrine mechanisms.16 26 27 In vivo limited dilution assay was conducted to evaluate the role of tumorous EHF on the crosstalk between PDACs and PSCs. As shown in figure 3A,B, the peritumorous injection of PSC-CM notably increased the tumour incidence in the PANC-1-vector group compared with that in the mice that received the control medium. However, the effect of PSC-CM was robustly suppressed in the PANC-1-EHF group.
PANC-1-vector, PANC-1-EHF, PANC-1-scramble and PANC-1-shEHF were incubated with the control medium or PSC-CM for 48 hours to determine whether EHF might regulate the crosstalk between PDAC cells and PSCs, and the proportions of CSCs were determined through flow cytometry. A PSC-CM stimulus could increase the proportion of CSC populations compared with those treated with the control medium (figure 3C–H). The abilities of PSC-CM to increase the CSC population were significantly enhanced by the short hairpin RNA knockdown of EHF and suppressed by the ectopic expression of EHF. Sphere formation and soft agar formation assays were also conducted. As shown in figure 3I–L, the promoting effects of PSC-CM on the self-renewal and anchorage-independent growth of CSCs were inhibited by the ectopic expression of EHF and remarkably increased by EHF depletion. These results were confirmed in multiple PDAC cell lines (online supplemental figures S5 and S6). Therefore, our results indicated that tumorous EHF decreased the sensitivity of PDACs to PSC stimulus.
Supplemental material
Supplemental material

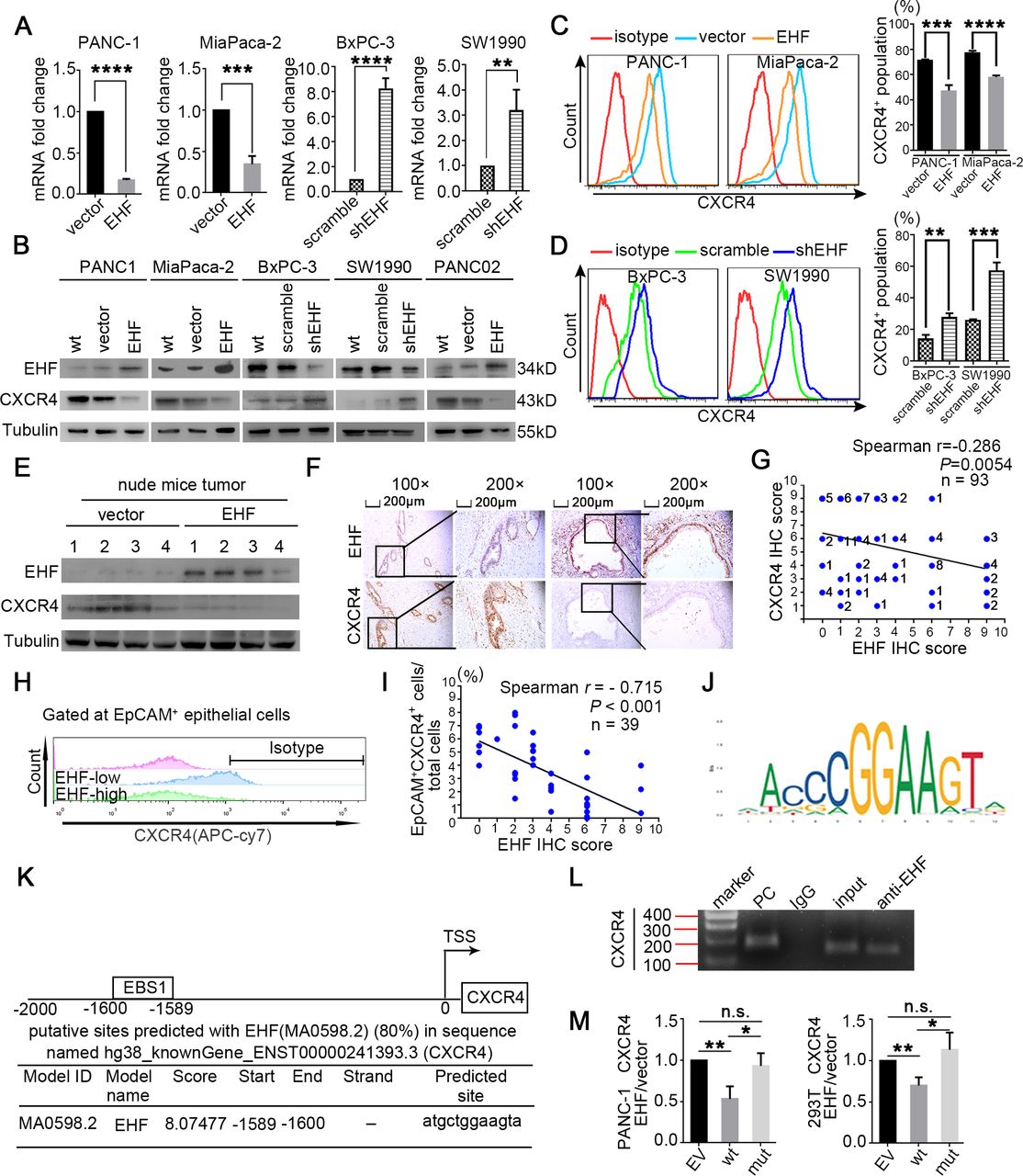
CXCR4 is transcriptionally repressed by EHF in PDAC. (A) Q-PCR on CXCR4 mRNA were performed in indicated cell lines. Actin was used as internal control. (B) Western blot on EHF and CXCR4 proteins in indicated cell lines was performed. Representative results are shown. (C,D) Percentages of CXCR4+ population in indicated cell lines were determined by flow cytometry. Representative histograms (C, left; D, left) and percentage of CXCR4+ population (C, right; D, right) are shown. (E) Western blot on EHF and CXCR4 proteins in harvested mice subcutaneous tumour tissues (tumour tissues were from figure 2I). β-Tubulin was used as loading control. Representative results are shown. (F,G) Representative IHC images of EHF and CXCR4 expression using human PDAC tissue sections (n=93) (F). Bars, 200 µm. Spearman rank correlation analysis was used to evaluate the correlation between tumorous EHF and CXCR4 expression (n=93) (G). The number at the right side of the plots represented the case number. (H,I) Single-cell suspensions were made from 39 cases of fresh PDAC tissues and stained with antibodies against CXCR4. Tumorous CXCR4+ cells were determined by flow cytometry. Gated on EpCAM+ cells to exclude non-epithelial cells. Representative histograms are shown (H). Spearman correlation analysis between EHF IHC score and the proportion of EpCAM+CXCR4+ population are shown (I); n=39, p<0.001. (J) EHF scanned motif logo. (K) Predicted EBSs on the human CXCR4 promoter. Position relative to the transcription start site of CXCR4, EBS sequence and corresponding JASPAR score. (L) Binding of EHF to the promoter of CXCR4 was determined by chromatin immunoprecipitation. IgG was used as negative control. Anti-RNA polymerase Ⅱ was used as positive control. Representative results are shown. (M) PANC-1 (left) and 293 T cells (right) were transfected with either vector control or pCDH-EHF in conjunction with the luciferase reporter pGL3-empty vector, pGL3-CXCR4-EBS1-wt or pGL3-CXCR4-EBS1-mut. Results were expressed as fold induction relative to that of the corresponding cells transfected with the control vector after normalisation of firefly luciferase activity according to Renilla luciferase activity. All experiments were repeated three times independently. Paired Student’s t-test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. EBS, EHF-binding site EHF, EHF, E26 transformation-specific homologous factor; IHC, immunohistochemistry; n.s., not significant; PC, positive control; PDAC, pancreatic ductal adenocarcinoma; Q-PCR, quantitative PCR; shEHF, short hairpin EHF; wt, wild type.

EHF decreases the sensitivity of PDACs to PSCs derived CSC-supporting stimulus by suppressing CXCR4. (A–C) BxPC-3-scramble/shEHF-scramble and BxPC-3-scramble/shEHF-shCXCR4 were cultured with PSC-CM or the Ctrl medium. The percentages of PCSCs in each cell line under each treatment are shown; the fold change of the percentage of PCSCs in each cell line after culturing with PSC-CM was calculated: (A) CD24+CD44+ cells, (B) ALDH+ cells, (C) CD133 +cells. representative dot plots/ histogram (left), the statistical analysis of CSC percentage of each group (medium) and the statistical analysis of the fold change in each cell line (right). (D) Statistical analysis of the sphere number of each cell line under the treatment of serum-free medium and serum-free medium with PSC-CM added (left), statistical analysis of the fold change of sphere number after culturing with serum-free medium containing PSC-CM in each cell line (right). (E) In vivo limited dilution assay was performed to determine the effects of PSC-CM on CSC self-renewal of BxPC-3-scramble/shEHF-scramble and BxPC-3-scramble/shEHF-shCXCR4.Tumour incidence and CSCs probabilities are shown. All experiments were repeated three times independently. Paired Student’s t-test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ALDH, aldehyde dehydrogenase; CM, conditioned medium; CSC, cancer stem cell; Ctrl, control; EHF, E26 transformation-specific homologous factor; n.s., non-significant; PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cell; SSC, side scatter.
Tumorous EHF abrogates the sensitivity of PDAC to PSC-derived CXCL12 stimulus
Blocking antibodies for cytokines secreted by PSCs were added to PSC-CM to identify the mechanism by which EHF regulated the PDAC–PSC crosstalk. CXCL12 was found to be the potential cytokine that induced the different reaction of PDACs to PSC-derived stimulus, depending on the EHF expression (online supplemental figure S7).
Supplemental material

Identification of compounds that induce EHF overexpression. (A,B). PANC-1, BxPC-3 and two primary cancer cell lines PDX1# and PDX2# were treated with rosiglitazone(5 and 10 µM, 24 hours). DMSO was used as control. (A) Q-PCR was conducted to detect for EHF mRNA expression. (B) Western blot for EHF expression was performed. Representative results are shown. (C) PPAR-γ-scanned motif logo (D) predicted PPREs on the human EHF promoter. position relative to the transcription start site of Ehf, PPRE sequences and corresponding JASPAR scores. (E) binding of PPAR-γ to the promoter of EHF was determined by chromatin immunoprecipitation. IgG was used as negative control. Anti-RNA PolymeraseⅡwas used as positive control. representative results were shown. (F) The promoter activity of EHF after treated with rosiglitazone. PANC-1 transfected with either luciferase reporter pGL3-empty vector or wild type pGL3-ESE3/EHF promoter were treated with rosiglitazone(10 µM,24h). Forty-8 hours later, cells were collected for dual luciferase assay. results were expressed as fold induction relative to those of the corresponding cells transfected with pGL3-empty vector after normalisation of firefly luciferase activity according to Renilla luciferase activity. All experiments were repeated three times independently. Paired Student’s t-test was used as statistical analysis. *p<0.05 and **p<0.01. DMSO, dimethyl sulfoxide; EHF, E26 transformation-specific homologous factor; PC, positive control; PPRE, PPAR-γ response element; PPAR-γ, peroxisome proliferator-activated receptor gamma;Q-PCR,quantitative PCR; TSS, transcriptional start site.
In in vivo limited dilution assay, CXCL12 notably increased the tumour incidence of PANC-1-vector group compared with that in the control medium group. However, the effect of CXCL12 was robustly suppressed in the PANC-1-EHF group (figure 4A,B). Then, the cell lines of PANC-1-vector, PANC-1-EHF, PANC-1-scramble and PANC-1-shEHF were treated with recombinant CXCL12 and the control medium. The CXCL12 treatment sharply increased the proportions of CSCs in low-EHF-expressing cell lines; the ectopic EHF overexpression remarkably suppressed the response to CXCL12 (figure 4C–H). Sphere formation and soft agar formation assays were also conducted. As shown in figure 4I–L, the CXCL12 treatment robustly increased tumour sphere formation and anchorage-independent growth, and the stimulating effects of CXCL12 were inhibited by the ectopic expression of EHF. Similar results were observed in other PDAC cell lines (online supplemental figures S8 and S9). Therefore, tumorous EHF abrogated the sensitivity of PDAC to the CXCL12 stimulus.
Supplemental material
Supplemental material

Rosiglitazone inhibits PDAC stemness and suppresses the sensitivity to the stemness-promoting stimulus by upregulating the EHF expression. (A,B) Adherent cells were pretreated with 5 µM rosiglitazone for 48 hours, and then cells were collected and cultured with serum-free medium containing 100 ng/mL human recombinant CXCL12 in low-adherent six-well plates. Representative results are shown (PANC-1, A; BxPC-3, B). (C) In vivo limiting dilution assays were performed to determine the effects of rosiglitazone on CSC self-renewal of PANC-1 cells with or without CXCL12 stimulus. Tumour incidences and CSCs probabilities are shown. (D) Rosiglitazone reduced the percentage of CXCR4+ population (5 µM, 24 hours). representative results are shown. (E) Schematic illustration for in vivo rosiglitazone therapeutic experiment in an orthotopic mice model. (F) Kaplan-Meier survival curves with log-rank test (PANC02-DMSO vs PANC02-rosiglitazone p=0.031). (G,H) Representative bioluminescent images of two groups on days 7 and 21 after tumour implantation (G). Statistical analysis of the fold change of BLI after drug treatment (BLI on day 21 to BLI on day 7; H) (n=8 per group). (I,J) Adherent PDAC-vector/EHF-KO cells were pretreated with 5 µM rosiglitazone for 48 hours; and then cells were collected and cultured with serum-free medium with or without 100 ng/mL human recombinant CXCL12 in low-adherent six-well plates for the following sphere formation assays. Sphere number analysis was shown (PANC-1, I; BxPC-3, J). (K) Schematic illustration for in vivo rosiglitazone therapeutic experiment using PANC02-vector/EHF-KO cell lines in an orthotopic mice model. (L,M) Representative bioluminescent images of the two groups on days 7 and 21 after tumour implantation are shown (L). Statistical analysis of the fold change of BLI after drug administration (BLI on day 21 to BLI on day 7, M) (n=8 per group). (N) Kaplan-Meier survival curves with log-rank test were used to analyse the different effect after treating with DMSO and rosiglitazone. All experiments were repeated three times independently. Paired Student’s t-test was used for statistical analysis for in vitro experiments and unpaired Student’s t-test was used for animal experiments. *P<0.05, **P<0.01, ***P<0.001, ****P<0.001. BLI, bioluminescent intensity; CSC, cancer stem cell; DMSO, dimethyl sulfoxide; EHF, E26 transformation-specific homologous factor; KO, knock out; n.s., non-significance; PDAC, pancreatic ductal adenocarcinoma.

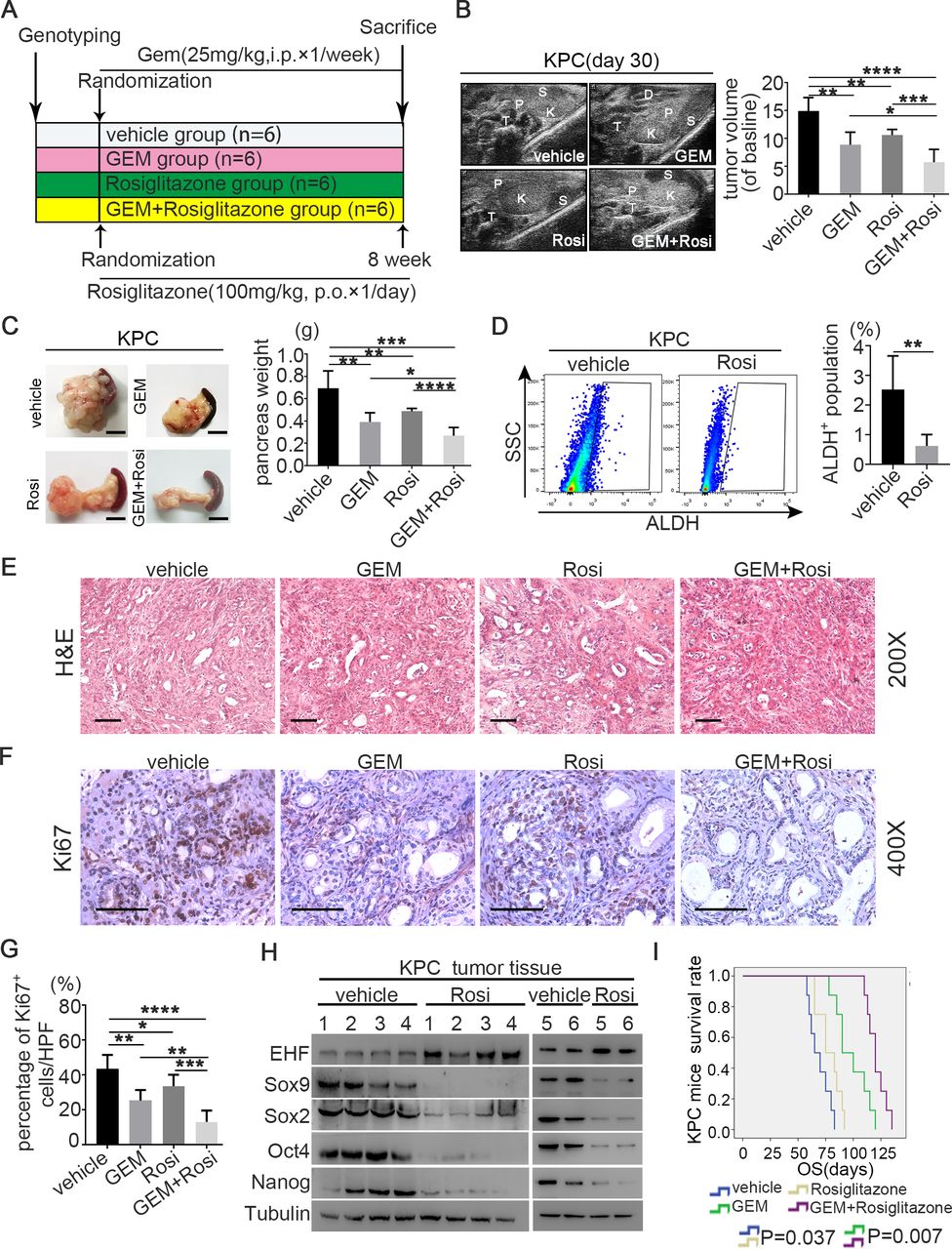
Rosi sensitises PDAC to gemcitabine therapy in theE KPC mouse model. (A) Experimental design programme. (B) Representative ultrasound images of KPC mice treated with vehicle (n=6), GEM (n=6), Rosi (n=6) and GEM+Rosi (n=6) at day 30 after drug treatment (left). Pancreatic T, P, S, K and D. Statistical analysis for the fold change of pancreatic T volumes measured by ultrasound system at day 30 after drug treatment (volumes in day 0 were used as baseline) (right). (C) Representative macroscopic images of pancreatic T in KPC mice treated with vehicle, GEM, Ros and GEM+Rosi after sacrifice (left). Statistical analysis for P weight of KPC mice from different groups (right). (D) Rosi reduced the percentage of ALDH+ population in PDAC in the KPC model. PI was used to exclude dead cells; CD45 was used to exclude leucocytes; and DEAB was used as negative control. Representative dot plots (left) and statistical analysis (right) are shown. (E) Representative images of H&E slides from tumours of four groups. Scale bars: 200 µm. (F) Representative images of KPC pancreatic tumour IHC for Ki-67 staining. Scale bars: 400 µm. (G) Statistical analysis for percentage of Ki-67-positive cells in different groups. (H) Protein expression of EHF and stemness markers (SOX9, Sox2, Nanog and Oct4) were detected in pancreatic T tissues of KPC mice by western blot. Tubulin was used as loading control. Representative results are shown. (I) Kaplan-Meier survival curves with log-rank test for KPC mice treated with vehicle (n=8), GEM (n=8), Rosi (n=8) and GEM+Rosi (n=8). The mouse experiments were repeated three times independently, and non-paired Student’s t-test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. ALDH, aldehyde dehydrogenase; D, duodenum; DEAB, diethylamino benzaldehyde; EHF, E26 transformation-specific homologous factor; GEM, gemcitabine; K, kidney; OS, overall survival; P, pancreas; PDAC, pancreatic ductal adenocarcinoma; PI, propidium Iodide; Rosi, rosiglitazone; S, spleen; SSC, side scatter; T, tumour.
CXCR4 is transcriptionally repressed by EHF in PDAC
CXCR4 and CXCR7 are the receptors of CXCL12. Tumorous EHF did not regulate the secretion of CXCL12 in PSCs in coculture experiments (online supplemental figure S10A,B), so we investigated whether EHF regulated the CXCR4/CXCR7 expression in PDACs. The expression of CXCR7 was not modulated by EHF (online supplemental figure S10C). Q-PCR, western blot and flow cytometry showed that the expression of CXCR4 was negatively regulated by EHF in the PDAC cell culture (figure 5A–D and online supplemental figure S11) and confirmed via western blot by using the harvested xenograft tumour tissues from the experiments in figure 2I (figure 5E). Importantly, the EHF expression was negatively correlated with the CXCR4 expression in human PDAC tumour tissues (figure 5F–I).
Supplemental material
Supplemental material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
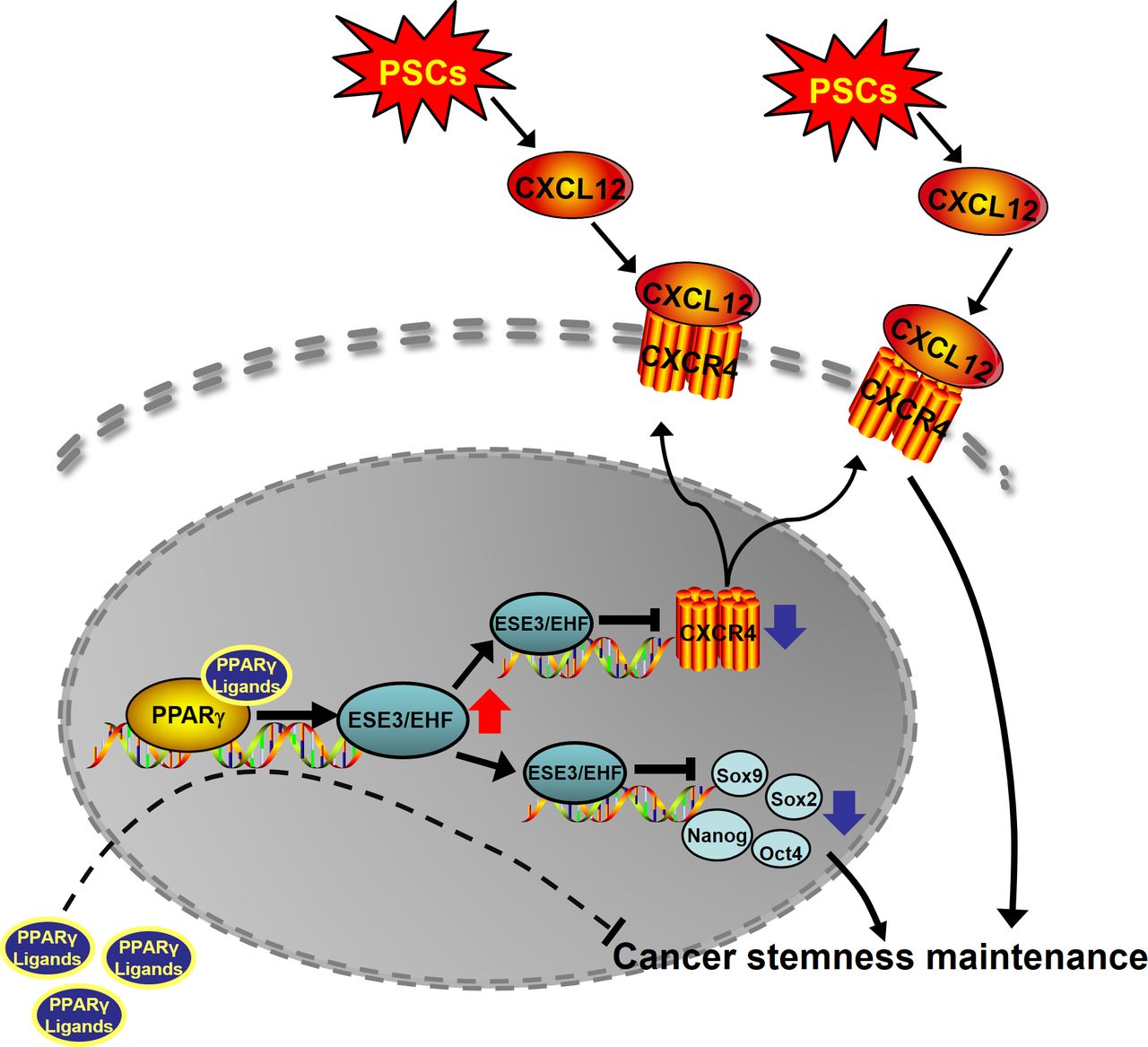
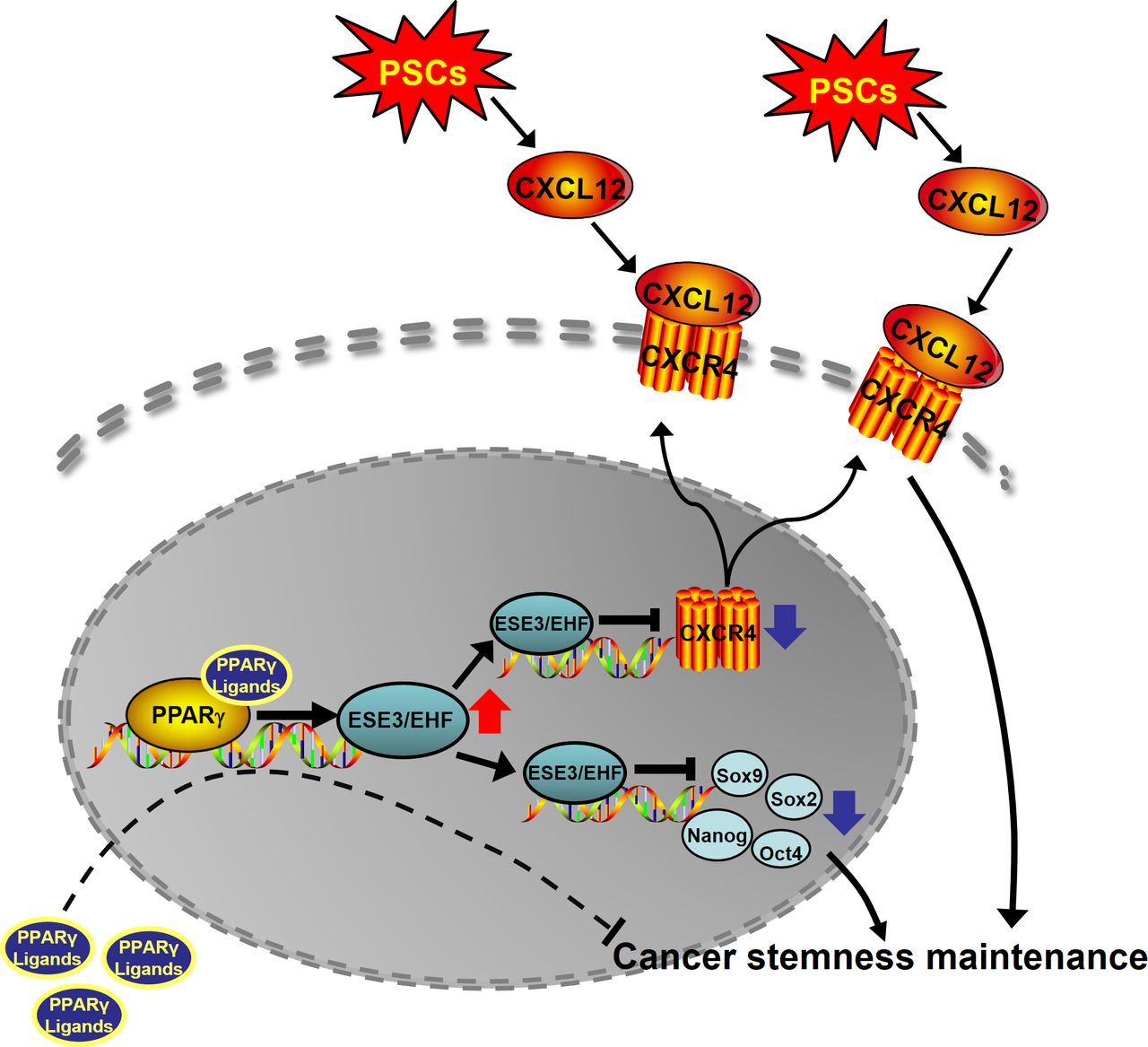
Schematic of the research. ESE3/EHF regulated PDAC CSCs property through cell-intrinsic and extrinsic pathway. Rosiglitazone suppressed PC stemness and inhibited the cross-talk between PC and PSCs by upregulating ESE3/EHF. CSC cancer stem cell; EHF, E26 transformation-specific homologous factor; PC, positive control; PDAC, pancreatic ductal adenocarcinoma; PPAR-γ, peroxisome proliferator-activated receptor gamma; PSC, pancreatic stellate cell.
In silico analysis showed one high-confidence EHF-binding site (EBS) within the promoter region of CXCR4 in the JASPAR database (figure 5J,K). ChIP was conducted in the PANC-1 cell line and revealed that EHF markedly bound to the promoter of human CXCR4 (figure 5L). The luciferase analysis of PANC-1 and 293T showed that the EHF overexpression significantly decreased the transcription of CXCR4 (PANC-1, p=0.0065; 293T, p=0.0061) and the mutation of EBS1 substantially abrogated the trans-suppression of the CXCR4 promoter induced by EHF (PANC-1, p=0.3295; 293T, p=0.4918; figure 5M).
EHF decreases the sensitivity of PDACs to PSC-derived CSC-supporting stimulus by suppressing CXCR4
BxPC-3-scramble-scramble, BxPC-3-scramble-shCXCR4, BxPC-3-shEHF-scramble and BxPC-3-shEHF-shCXCR4 were established (online supplemental figure S12A,B) and treated with the control medium or PSC-CM to evaluate whether EHF decreased the sensitivity of PDACs to PSC-CM stimulus by suppressing CXCR4. As we previously observed, EHF depletion with shRNA increased the pro-CSC effects of PSC-CM (figure 6A–C). However, knocking down CXCR4 in BxPC-3-shEHF cells almost abrogated the effects of PSC-CM treatment (figure 6A–C). Similarly, the shRNA depletion of CXCR4 in PDAC cells abrogated the abilities of PSC-CM to promote the sphere formation of PDAC tumour (figure 6D). In vivo limited dilution assay demonstrated that PSC-CM significantly increased the CSC frequency in PDAC cells (from 1/779769 to 1/245406), and the effects of PSC-CM were dramatically enhanced after EHF knockdown (CSC frequency increased from 1/210828 in the 1640 group to 1/1443 in the PSC-CM group; figure 6E). Strikingly, CXCR4 knockdown abrogated the pro-CSC effects of the conditioned medium from PSC (figure 6E). Collectively, our data supported that tumorous EHF decreased the sensitivity of PDACs to PSC-derived CSC stimulus by suppressing the CXCR4 expression.
Supplemental material
Identification of compounds that induce EHF overexpression
EHF is a promising therapeutic target of PDAC, so 190 compounds from a drug library in our laboratory were screened to determine their effects on regulating the EHF expression (online supplemental table S3 and online supplemental figure S13A). Among these 190 compounds, 14 could induce the EHF overexpression (online supplemental figure S13B). Considering the efficacy on the EHF upregulation and the safety profile, we chose rosiglitazone as a candidate for further studies. As shown in figure 7A,B, rosiglitazone significantly induced the mRNA and protein expression of EHF in PDAC cells and PDX-derived PDAC cells in a concentration-dependent manner.
Supplemental material
PPAR-γ is a ligand-activated nuclear transcription factor. Rosiglitazone is a specific PPAR-γ agonist that improves glycaemic control and insulin sensitivity in patients with diabetes by selectively activating PPAR-γ. Computational analysis showed two high-confidence PPAR-γ response elements (PPREs) corresponding to the promoter regions of EHF in the JASPAR database (figure 7C,D). ChIP primers were designed to investigate the binding site through the ChIP assay and evaluate whether PPAR-γ directly bound to the promoter of EHF. As shown in figure 7E, PPAR-γ antibody could precipitate the PPRE sequence, which indicated the direct binding of PPAR-γ to the EHF promoter. Luciferase analysis showed that rosiglitazone significantly increased the transcriptional activity of the EHF promoter (p=0.0423), suggesting that rosiglitazone induced the EHF overexpression through PPAR-γ activation (figure 7F).
Rosiglitazone inhibits PDAC stemness and suppresses the sensitivity to the stemness-promoting stimulus by upregulating the EHF expression
As shown in figure 8A,B, rosiglitazone treatment inhibited the sphere formation capacity of PDACs and essentially blocked the CXCL12-mediated increase in the sphere formation of PDAC tumour. This finding suggested that rosiglitazone could be used to abrogate CXCL12 and PSC-mediated PDAC CSC self-renewal. To rigorously evaluate this hypothesis, we performed in vivo limited dilution tumourigenicity assay. As shown in figure 8C, rosiglitazone significantly reduced tumour initiation and CSC frequency. Moreover, no significant difference in tumour incidence could be observed in the PANC-1-CXCL12-rosiglitazone and PANC-1-ctrl-rosiglitazone groups. This result indicated that the stemness-promoting effect of CXCL12 was blocked by rosiglitazone. We also observed that rosiglitazone significantly deceased the percentage of the CXCR4+ population in PDAC cells (figure 8D). In the orthotopic BALB/C tumour mouse model, the mice in the rosiglitazone group survived significantly longer than those in the dimethyl sulfoxide (DMSO) control group (p=0.031; figure 8E,F). The normalised BLI in the rosiglitazone group was notably lower than that in the DMSO control group, suggesting that orthotopic tumour growth was inhibited (figure 8G,H). The harvested tumours from the orthotopic mouse model were analysed throughflow cytometry. As shown in online supplemental figure S14A, rosiglitazone significantly decreased the proportion of ALDH+ cells. Western blot and IHC indicated that rosiglitazone increased the expression of EHF and decreased the expression of stemness genes (online supplemental figure S14B,C).
Supplemental material
EHF was knocked out in PANC-1, BxPC-3 and PANC02 cells via the CRISPR/dCas9 system to determine whether rosiglitazone inhibited the stemness of PDACs via the PPAR-γ-EHF pathway (online supplemental figure S15). As shown in figure 8I,J, EHF-KO could abrogate the abilities of rosiglitazone to inhibit tumour sphere formation. In the orthotopic tumour mouse model (figure 8K–M), rosiglitazone significantly reduced the tumour burden in the PANC02-vector group but not in the EHF-KO group. Importantly, rosiglitazone could improve survival in the vector control group but not in the EHF-KO group (figure 8N).
Supplemental material
Ibuprofen and allopurinol, two other compounds that upregulated the EHF expression, could similarly suppress CSC stemness in PDAC (online supplemental figures S16 and S17).
Supplemental material
Supplemental material
Rosiglitazone sensitises PDAC to gemcitabine therapy in the KPC mouse model
Given the role of CSCs in chemotherapy resistance and the function of rosiglitazone on suppressing PDAC stemness, KPC mouse models were used to evaluate the therapeutic effects of gemcitabine plus rosiglitazone. When the tumour volumes reached 20–60 mm3, the KPC mice were randomised into four groups: vehicle, GEM, rosiglitazone and GEM plus rosiglitazone groups (figure 9A and online supplemental figure S18). Ultrasonic imaging showed that the tumour burdens significantly decreased in the GEM plus rosiglitazone group compared with those in the GEM group alone on day 30 (figure 9B). Consistently, GEM plus rosiglitazone reduced the weight of pancreas compared with that of GEM monotherapy (figure 9C). The proportion of ALDH+ cells significantly decreased when rosiglitazone was administered (figure 9D,E). The IHC of ki-67 indicated that GEM plus rosiglitazone combination therapy more significantly reduced cancer cell proliferation compared with that with GEM monotherapy (figure 9F,G). The western blot of pancreatic tumour tissues from the KPC mice further confirmed that rosiglitazone could induce the expression of EHF and suppress the expression of stemness marker genes (figure 9H). Finally, obvious survival benefits were observed in the GEM plus rosiglitazone group compared with those in the GEM group (figure 9I). Therefore, rosiglitazone suppressed PC stemness and could be used as a new therapeutic method in the clinical practice of PDAC treatment (figure 10).
Supplemental material
Discussion
CSCs play a significant role in disease recurrence and treatment failure in PC. The crosstalk between CSCs and its niche is essential for stemness maintenance, cancer initiation and progression.3 28 Seino et al 29 revealed that cancer-associated fibroblasts transmit a protumourigenic niche signal to PDACs through the juxtacrine production of stromal Wnt ligands. Öhlund et al 30 reported that tumour organoids need PSC-secreted ligands for surviving. Our study supported that the cytokines IL-6, IL-8 and CXCL12 secreted by PSCs significantly increase the cancer stemness of PDAC.30–32 A PSC-derived CSCs niche shows potential for applications that promote cancer stemness, so targeting the crosstalk between CSCs and PSCs can be an efficient modality for the prevention of tumour recurrence.
EHF is a member of a highly diverse ETS superfamily. Our group first demonstrated EHF as a tumour suppressor that directly inhibits PDAC progression by upregulating E-cadherin while downregulating TGF-β1 and GM-CSF.19 20 The role of EHF in CSCs regulation was first identified in prostate cancer. Albino et al 33 reported that EHF directly controls the activity of the Lin28/let-7 axis, a key pathway involved in CSC expansion. EHF also represses the expression of the key CSCs genes TWIST1, ZEB2, BMI1 and POU5F1.23 Furthermore, the loss of EHF leads to the upregulation of IL-6 and the activation of the JAK/STAT3 pathway.21 Our data indicated that EHF not only plays a cell autonomous role in regulating CSC stemness but also has important functions in regulating the sensitivity to a pro-CSC stimulus from the PSC niche.
PSCs within the tumour microenvironment represent the principal source of CXCL12, which binds to its two receptors, CXCR4 and CXCR7, to activate a number of signalling pathways in PC cells, such as the PKCα/NFκB, MAPK, PI3K-Akt-mTOR and JAK/STAT pathways.34 35 Moreover, Hermann et al 6 defined a subpopulation of migrating CSCs that are characterised by the expression of the CXCR4 receptor and critically involved in tumour metastasis. Khan et al 16 reported that CXCR4/CXCL12 and hedgehog signalling pathways mediate the chemoresistance of PC cells on coculturing with PSCs. We found that tumorous EHF repressed the CXCR4 expression but not the CXCR7 expression. ChIP and dual-luciferase assays revealed that EHF directly bound to the promoter regions of CXCR4 to suppress its transcription. Our blocking experiment revealed that EHF decreased the sensitivity of cancer cells to the PSC stimulus by repressing the CXCR4 expression.
Our findings suggested that restoring the CSC-suppressing functions of EHF could be a promising approach in PDAC treatment. Here, we screened 190 compounds from our drug library on the basis of their effects on the EHF expression. These 190 compounds are commonly used drugs in clinical work and easily obtained. Rosiglitazone, a high-affinity synthetic agonist for nuclear PPAR-γ, was identified as the most potent activator of the EHF expression with limited side effects. On activation with specific ligands, PPAR-γ binds to PPREs, which then mediate the target gene expression.36 Recently, studies have indicated that rosiglitazone and related thiazolidinediones may play inhibitory roles in various types of cancer cells, including PC, such as enhancing radiosensitivity,37 reducing immune suppression38 and inhibiting cell invasion and metastasis.39–41 In our current work, rosiglitazone-activated PPAR-γ bound to the promoter region of EHF and upregulated its expression. In vivo and in vitro studies demonstrated that rosiglitazone decreased the sensitivity of PDAC to PSCs’ stimulus and inhibited tumour stemness properties by inducing tumorous EHF expression. Our results suggested that the effects of rosiglitazone on EHF upregulation could be translated into the development of targeted therapy against cancer stemness.
Our study first reported that EHF suppressed cancer stemness from intrinsic and extrinsic pathways. For the intrinsic pathway, EHF repressed the expression of SOX9, SOX2, OCT4 and Nanog. For the extrinsic pathway, EHF decreased the sensitivity of PDACs to the stimulus from the PSC-derived CSC-supportive niche by negatively regulating the tumorous CXCR4 expression. Conceivably, rosiglitazone could be used to target pancreatic stem cells and the crosstalk between CSCs and its niche by upregulating EHF.
Materials and methods
Patient and sample collection
A total of 93 sequential PDAC tissues were retrospectively collected from patients who received radical surgery R0 resection at the Tianjin Medical University Cancer Institute and Hospital from July 2011 to January 2015. The follow-up rate was 100% until the last follow-up on 23 October 2019. Then, 39 consecutive cases of fresh PDAC tissues were prospectively collected during operation from January 2018 to November 2019.
Primary human PC cells
Human pancreatic tumours were obtained during surgery with written informed consent from all the patients. Low-passage (<10 passages) primary cancer cells were used for later experiments. Information of patients and cellular genomic background was listed in online supplemental tables S4, S5.
Cell culture and transfection
The PC cell lines PANC-1, MiaPaca-2, BxPC-3, SW1990 and PANC02 were maintained as previously described.20 42 The human PSCs were set up as reported by Jesnowski et al.43 PDAC-vector, PDAC-EHF, PDAC-scramble and PDAC-shEHF were established as previously described, and related sequence information is listed in online supplemental table S6.20 Mycoplasma contamination was excluded in these cell lines. The cells were cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2 with Dulbecco’s Modified Eagle Medium (DMEM) and Roswell Park Memorial Institute-1640 (RPMI-1640) basic medium supplemented with 10% fetal bovine serum (FBS) as a medium.
Animals
Female NOD/SCID, BALB/C nude and KPC mouse models (4–6 weeks old) were used. All the mice were maintained in specific pathogen-free conditions.
In vivo tumourigenicity assay
The cohorts of NOD/SCID mice were randomised into different groups. In each group, cancer cells at different dilutions were subcutaneously injected into the contralateral flanks of the NOD/SCID mice. Stem cell frequency was calculated via the website (http://bioinf.wehi.edu.au/software/elda/).
Orthotopic mouse model
The cohorts of BALB/C nude mice were randomised into different groups. An orthotopic model was established using 5×105 luciferase-expressing PDAC tumour cells. Tumour growth was analysed through BLI.
KPC mouse model and preclinical animal cohorts
LSL-KrasG12D/+, LSL-Trp53R172H/+ and Pdx1-Cre mouse models were were generated in-house. Primers and PCR conditions for genotyping were listed in online supplemental tables S7, S8. Preclinical studies were conducted with a KPC mouse model.
Immunohistochemistry (IHC) and multiplex fluorescent IHC
IHC score was determined by two independent pathologists who were blinded to the patients’ clinicopathological features and prognosis. For multiplex fluorescent IHC, stained tissues were scanned and captured using a Vectra Polaris system (PerkinElmer). Images captured were analysed using the inForm cell analysis software (PerkinElmer).
Flow cytometry
Primary pancreatic cells, PC cell lines and cells from tumour tissue digestions were stained with anti-hCD133, anti-hCD24, anti-hCD44, anti-ESA, anti-hCXCR4 or appropriate control antibodies. Detailed information of antibodies used is listed in online supplemental table S9. The ALDH activity was detected with ALDEFLUOR kits. Isotype controls were used as negative controls. Data were analysed using FlowJo V.10.0.
Reverse transcription PCR (RT-PCR)
The total RNA of the cells was extracted with TRizol and converted to cDNAs by using an RT-PCR system. Then, real-time fluorescent Q-PCR was conducted to analyse the cDNA levels. Related primers are listed in online supplemental table S10.
Western blot
Target proteins were detected through western blot with primary antibodies as follows: anti-EHF, anti-Sox9, anti-Sox2, anti-Nanog, anti-Oct4, anti-CXCR4, anti-E-cadherin, anti-CK19, anti-CAII and anti-tubulin.
Anchorage-independent growth assay
Each six-well plate was coated with 1 mL of bottom agar, and 5000 cells were suspended in 1 mL of the top agar. Cells were incubated for 21 days, and colonies were analysed.
Sphere formation assay
Cells (5000 cells/mL) were cultured in ultralow adhesion plates in a serum-free medium. After the cells were cultured for 2 weeks, tumour spheres with a diameter of >75 µm were counted.
ChIP and luciferase analysis
ChIP assays were performed using a ChIP kit. The immunoprecipitated products were detected through PCR assays. Luciferase analysis was performed on the basis of the binding sites identified through ChIP analysis. Related sequences are listed in online supplemental table S11.
Preparation of PSC-CM
PSCs were grown to 70%–80% confluence in 10 cm dishes in complete culture media. Then, the medium was replaced with FBS-free DMEM/F12 (1:1), and the cells were cultured for additional 48 hours.
Stimulation of PSC-CM/CXCL12
For in vitro studies, PSC-CM was added to the culture medium at a ratio of 1:1, and CXCL12 was added to the culture system at a final concentration of 100 ng/mL. For in vivo studies, 200 µL of PSC-CM or 200 µL of CXCL12 was intratumourally injected three times a week.
Treatment of rosiglitazone
For in vitro studies, adherent PDAC cells were pretreated with 5 µM rosiglitazone for 48 hours and were collected for further experiments. For in vivo tumourigenicity studies, rosiglitazone (100 mg/kg/day) was peritumorously injected three times a week. For orthotopic tumour models, rosiglitazone (100 mg/kg/day) was injected intraperitoneally three times a week.
Statistical analysis
Statistical analysis was performed with IBM SPSS Statistics V.21.0. Each experiment was conducted in triplicate, and data were presented as mean±SD unless otherwise stated. The variance between the groups was statistically compared. Student’s t test was conducted to compare the mean values. Kaplan–Meier curves were analysed for median survival. A log-rank test was carried out to analyse the differences in survival times among the patient subgroups. *P<0.05, **P<0.01 and ***P<0.001 indicated significant differences, and n.s. meant non-significant. See online supplemental Methods for details.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
All the patients provided written consent for the use of their specimens and disease information for future investigations according to the ethics committee of Tianjin Medical University Cancer Institute and Hospital, China, and in accordance with the recognised ethical guidelines of Helsinki (ID number of the ethics approval: Ek2017141). Animal experiment procedures were approved by the ethics committee of Tianjin Medical University Cancer Institute and Hospital, in compliance with the principles and procedures of the NIH Guide for the Care and Use of Laboratory Animals.
Acknowledgments
We thank Professor Xueli Bai and Professor Qi Zhang (Department of Surgery, the Second Affiliated Hospital, Zhejiang University, China) for their technical assistance.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1
- Data supplement 2
- Data supplement 3
- Data supplement 4
- Data supplement 5
- Data supplement 6
- Data supplement 7
- Data supplement 8
- Data supplement 9
- Data supplement 10
- Data supplement 11
- Data supplement 12
- Data supplement 13
- Data supplement 14
- Data supplement 15
- Data supplement 16
- Data supplement 17
- Data supplement 18
- Data supplement 19
- Data supplement 20
Footnotes
TZ, JL and YX contributed equally.
Contributors JH and JL conceived and designed the experiments; TZ and YX performed most of the experiments; JL, WB, KZ, WJ, SY, HoWang and HaWang performed some experiments; YG, CH and SY provided technical support; TZ, SG, XW provided patient samples and technical support. JH, JL, TZ and YX analysed and discussed the data. JL and TZ wrote and completed the paper. JH supervised the entire project.
Funding This work was supported by the National Natural Science Foundation of China (grants 82030092, 81720108028, 82072657, 81802432, 82072716, 81802433, 82072659, 81871968 and 81871978), the programmes of Tianjin Prominent Talents, Tianjin Eminent Scholars, Tianjin Natural Science Fundation (20JCQNJC01330, 18JCJQJC47800, 19JCJQJC63100 and 19JCYBJC26200), Tianjin Postgraduate Research and Innovation Project (2019YJSB104), and Tianjin Research Innovation Project for Postgraduate Students (2019YJSB104). The research in S. Yang’s laboratory is supported by the National Cancer Institute (R01 CA175741) and the Elsa U. Pardee Foundation (R01 CA175741)
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.