Article Text

Abstract
Background Worldwide, gastric cancer (GC) is the fourth most common malignancy and the most common cancer in East Asia. Development of targeted therapies for this disease has focused on a few known oncogenes but has had limited effects.
Objective To determine oncogenic mechanisms and novel therapeutic targets specific for GC by identifying commonly dysregulated genes from the tumours of both Asian-Pacific and Caucasian patients.
Methods We generated transcriptomic profiles of 22 Caucasian GC tumours and their matched non-cancerous samples and performed an integrative analysis across different GC gene expression datasets. We examined the inhibition of commonly overexpressed oncogenes and their constituent signalling pathways by RNAi and/or pharmacological inhibition.
Results Hepatocyte nuclear factor-4α (HNF4α) upregulation was a key signalling event in gastric tumours from both Caucasian and Asian patients, and HNF4α antagonism was antineoplastic. Perturbation experiments in GC tumour cell lines and xenograft models further demonstrated that HNF4α is downregulated by AMPKα signalling and the AMPK agonist metformin; blockade of HNF4α activity resulted in cyclin downregulation, cell cycle arrest and tumour growth inhibition. HNF4α also regulated WNT signalling through its target gene WNT5A, a potential prognostic marker of diffuse type gastric tumours.
Conclusions Our results indicate that HNF4α is a targetable oncoprotein in GC, is regulated by AMPK signalling through AMPKα and resides upstream of WNT signalling. HNF4α may regulate ‘metabolic switch’ characteristic of a general malignant phenotype and its target WNT5A has potential prognostic values. The AMPKα-HNF4α-WNT5A signalling cascade represents a potentially targetable pathway for drug development.
- GASTRIC CANCER
- DRUG DEVELOPMENT
- ONCOGENES
- MOLECULAR BIOLOGY
- GENE EXPRESSION
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Gastric cancer is the fourth leading cause of cancer death in the world, having an estimated 989 000 new cases of cancer annually, with 738 000 deaths.
While several risk factors are known (including Helicobacter pylori infection, dietary factors and gastric reflux), targeted therapies remain largely undeveloped.
What are the new findings?
Integrative analysis of Caucasian and Asian-Pacific gastric tumour expression datasets (including newly generated transcriptomic profiling of 22 tumours in this study) revealed a relatively small common set of highly overexpressed genes. Of those genes, hepatocyte nuclear factor-4α (HNF4α) was one key transcription factor.
Inhibitory RNA and pharmacological inhibition of HNF4α demonstrated antineoplastic activity in vitro and in vivo via downregulation of cyclins, cell cycle arrest and apoptosis.
In agreement with HNF4α being a known substrate that is downregulated by the AMPK energy-sensing kinase, the AMPK agonist metformin demonstrated an antitumor effect similar to direct HNF4α antagonists.
WNT5A is an HNF4α target gene, thus suggesting that an AMPK–HNF4α–WNT oncogenic signalling axis is likely to be involved in gastric tumour metabolism.
How might it impact on clinical practice in the foreseeable future?
Development of therapies that specifically target the AMPK–HNF4α–WNT signal cascade would probably represent effective approaches with minimal toxic effects for the management of gastric cancer.
Introduction
Gastric cancer (GC) is the fourth most common type of cancer in the world, with 989 000 new cases (7.86% of total global cancers) annually, and 738 000 deaths (9.7% of total global cancer deaths).1 In East Asia, GC is the most common type of cancer2 ,3; in North America and Europe, while the overall incidence of GC has been decreasing, the prevalence of proximal GC has increased notably.4 Biologically, gastric adenocarcinomas have been largely associated with two infectious agents, Helicobacter pylori5 ,6 and Epstein–Barr virus,7 and hereditary diffuse GC has been associated with germline mutations of cadherin-1 (CDH1).8 ,9
Surgical removal of early stage GC tumours is key to effective treatment, but because few symptoms accompany early GC, most tumours are found at an advanced stage.10 GC recurrence rates are high among all populations, and although improved early detection has increased the 5-year survival rate by 67% in Korea11 and 57% in Japan,12 the global 5-year survival rate has remained at 20–30% for the past 35 years.10 Efforts to develop GC-targeted therapies have focused on a few well-known oncogenes. For example, one targeted therapy, trastuzumab, was approved for use in combination with chemotherapy as a first-line treatment of ERBB2-positive advanced and metastatic GC,13 ,14 and several other targeted therapies remain under investigation.15–17 However, there remains an urgent need to identify novel targetable oncogenic mechanisms.18–20
To develop more effective GC-specific therapies, we (1) generated whole-transcriptome profiles for GC in Caucasians and identified hepatocyte nuclear factor-4α (HNF4α) as a key component across multiple GC expression datasets; (2) demonstrated that HNF4α knockdown was anti-tumorigenic in both cell line and animal studies; (3) elucidated the regulatory context of HNF4α by showing it acts downstream of AMP-activated protein kinase (AMPK) and upstream of wingless-type MMTV integration site (WNT) signalling; and (4) showed the prognostic value of WNT5A, a HNF4α downstream target gene, in patients with GC. Collectively, our study reveals a critical role of AMPKα-HNF4α-WNT5A signalling in the pathogenesis of GC, representing a potential therapeutic target.
Materials and methods
Sample collection and characterisation
For RNA-seq experiments, tumours were collected from a retrospective study using archival fresh-frozen human tissue specimens obtained from consented patients at the University of Texas MD Anderson Cancer Center Research Resource Tissue Bank (Houston, Texas, USA), as approved by the Institutional Review Board of MD Anderson Cancer Center. Histological classification and tumour stage were determined by a pathologist in the Department of Pathology at MD Anderson. Our analysis included 22 gastric tumours and their matched non-tumour tissues obtained from Caucasian patients who met our study criteria (sufficient amount and quality of RNA). Clinical and histopathological patient characteristics are summarised in online supplementary table S1.
Tissue microarrays were created from tissue samples from patients with GC who underwent gastrectomy and extended lymphadenectomy in 2005 and 2006 at the National Cancer Center in the Republic of Korea, as approved by the Institutional Review Board of the National Cancer Center in the Republic of Korea. Exclusion criteria were as follows: pT1; no R0 resection; distant metastasis (including peritoneal cytology-positive) present before or at the time of surgery; recurrent cancer; neoadjuvant therapy; cancer in remnant stomach; unusual histology other than papillary, tubular, signet ring cell or mucinous type; incomplete follow-up; paraffin-embedded tissues unavailable for tissue microarray; or other cancer history. These exclusions resulted in final datasets of 502 samples (discovery set: 251 cases in 2006, validation set: 251 in 2005). All patients had undergone D1+β or more lymph node dissection. Tumour stages were reclassified according to the 7th edition of the tumour staging system of the Union for International Cancer Control. Case characteristics (see online supplementary table S3) were determined and reviewed by a pathologist in the National Cancer Center Pathology Division.
To validate loss of AMPKα2 expression, 10 patient tissue samples per tumour stage were provided as a gift from Dr Hyun Cheol Chung, Cancer Center, Yonsei University Severance Hospital, Republic of Korea.
Experimental procedure for RNA-seq
RNA-seq was conducted at the Sequencing and Non-coding RNA Core Facility at MD Anderson Cancer Center. RNA isolation, library preparation (paired-end, 50nt and 35nt), template bead preparation and SOLiD V.4.0 sequencing followed standard protocols provided by Life Technologies (Carlsbad, California, USA), similar to our previous study.20 Sequencing data were submitted to the NCBI Gene Expression Omnibus.
Differential expression analysis
To analyse RNA-seq data from Caucasian patient GC tumours, we mapped short reads to the human reference genome (hg19) and exon junctions (defined as RefSeq gene annotation), using the whole-transcriptome analysis pipeline in the Life Sciences Bioscope (V.1.2 Life Technologies) with default parameters. We included only properly mapped read pairs and quantified gene expression levels as reads per kilobase of exons per million mapped reads (RPKM), log-transformed. We used paired t tests to detect genes differentially expressed between GC and normal samples, with significance at p<0.05. We obtained another three published GC gene expression datasets, two from the NCBI Gene Expression Omnibus: GSE13861 and GSE3696820 21 and one from TCGA22 (level 3 data obtained from TCGA data portal). We used t tests to detect differentially expressed genes and p<0.0001, p<0.05 and p<0.001 as the respective cut-offs. For 396 genes commonly identified across the four differential analyses, we classified them into five groups based on the directional change (upregulation/downregulation relative to normal) and the average fold change: (1) consistent upregulation with >2 fold; (2) consistent upregulation with <2 fold; (3) consistent downregulation with >2 fold; (4) consistent downregulation with <2 fold; and (5) inconsistent change.
Experiments on biological functions
Cells and reagents
We obtained human NCI-N87, AGS and HS 746T tumour cells from the American Type Culture Collection (Manassas, Virginia, USA). The MKN45 GC cell line was made available from Yonsei Cancer Center (Seoul, Korea). SNU-1–5, -16, -620, -216, -484,-601, -638, -668 and SNU-719 GC cell lines were made available from the Korean Cell Line Bank (http://cellbank_snu.ac.kr/). Monoclonal antibodies to human hypoxia-inducible factor 1α (HIF-1α) and β-catenin were purchased from BD Transduction Laboratories (San Jose, California, USA); AMPKα, LKB1, HNF4α, antiapoptotic antibody kit (tPARP, cPARP, t-caspase 3, c-caspace 3), t-mTOR, p-mTOR (Ser 2448 and Ser2481), raptor, eIF4E, S6,4E-BP1, WNT1, WNT3, GSK, β-catenin, TCF, LEF, pCAMKII, cyclin A, cyclin B, cyclin D1, cyclin D2 and β-actin were purchased from Cell Signaling Technologies (Boston, Massachusetts, USA) or Santa Cruz Biotechnology (Santa Cruz, California, USA). Antibodies against WNT5A, GSK, β-catenin, pCAMKII, cyclin D2, cyclin D1, cyclin A and cyclin B were purchased from Abgent (San Diego, California, USA), and metformin and puromycin were purchased from Sigma-Aldrich (St Louis, Missouri, USA). SBI-0634689.0001 (BI6015, an HNF4α antagonist) was provided by the Sanford-Burnham Medical Institute (La Jolla, California, USA).
Cell culture
The following human GC cell lines were used within 6 months of tissue resuscitation: NCI-N87, AGS, HS 746T, MKN45, SNU-1 SNU-5, SNU-16, SNU-620, SNU-216, SNU-484, SNU-601, SNU-638, SNU-668 and SNU-719, and cultured in RPMI-1640 (CellGro, Herndon, Virginia, USA) and 10% fetal calf serum (Hyclone, Logan, Utah, USA) at 37°C in 5% CO2. Cell line validation was by short tandem repeat profiling (ATCC). Cells (2.5×105) were seeded and grown under normoxic conditions to 70–80% confluence and then incubated in the presence or absence of 10 mM metformin for 0–5 days.
Western blotting
Cells grown under normoxic conditions in the presence or absence of 10 mM metformin were washed twice in phosphate-buffered saline and western blotting was conducted as previously described.23 Antibody-bound blots were quantified using ImageQuant software (Molecular Dynamics/GE Healthcare Biosciences, Sunnyvale, California, USA).
Quantitative RT-PCR
Total RNA was isolated from cell lysates using PARIS kits (Ambion/Applied Biosystems, Foster City, California, USA) according to the manufacturer's protocol. Next, quantitative RT-PCR was performed using an ABI 7300 system and TaqMan one-step Master Mix kit and predesigned primer/probe pairs for AMPKα1, AMPKα2, AMPKβ1, AMPKβ2, AMPKγ1 AMPKγ2, AMPKγ3 LKB1, HNF4α and β2-microglobulin (Applied Biosystems). Relative expression levels were normalised to β2-microglobulin using the 2(−delta-delta C(T)) method24 and Applied Biosystems GeneAmp 5700 SDS software. All measurements were performed in triplicate.
Small interfering RNA transfection
Small interfering RNA (siRNA) SMARTpool (set of four oligonucleotides) (Dharmacon/Thermo Fisher Scientific (Waltham, Massachusetts, USA) were used to transfect cells at 25 nM siRNA-HNF4α or a siRNA scrambled control, using Dharma-FECT 1 lipid transfection reagent. The transfection medium was removed after 24 h and replaced with fresh medium and the cells then grown in 5% CO2 at 37°C for an additional 48–72 h. RT-PCR and/or western blot analyses were performed to confirm target knockdown by siRNA.
Short hairpin RNA silencing
Short hairpin RNAs (shRNAs) (Sigma-Aldrich) were used to stably infect NCI-N87 GC cells (shRNA empty vector or HNF4α) plated in 12-well plates and maintained in RPMI 1640 plus 10% fetal bovine saline (FBS), with selection by 0.3 mg/mL puromycin.
Cell death assays
Five GC cell lines (AGS, N87, NCC19, NCC59 and SNU1967) were grown under the conditions described above. For assaying autophagy, a Cyto-ID Autophagy detection kit (Enzo Life Sciences, Farmingdale, New York, USA) was used according to the manufacturer's protocol. For assaying apoptosis, an Annexin V: PE Apoptosis detection Kit I (BD Biosciences, Franklin Lakes, New Jersey, USA) was used according to the manufacturer's protocol. FACSVerse (BD Biosciences) was used for flow cytometry analysis.
Reporter (Luciferase) assay
In the study, 96-well plates were seeded with 20 000 cells/well and transiently transfected with 25 nM scrambled or HNF4α siRNA. TCF/LEF-responsive luciferase construct, HNF4α, TCF/LEF, negative control and positive control genes were transfected, respectively (QIAGEN Sciences, Gaithersburg, Maryland, USA). Transfected cells were then treated with 10 μM metformin for 24–72 h. The positive control was a constitutively expressed green fluorescent protein (GFP) construct and the negative control was a minimal promoter GFP reporter. This assay was performed on MKN45, NCI-N87, NCC19, NCC59, NCC24 and SNU1967 GC cells. After transfection for 24, 48 and 72 h, cells were lysed with passive lysis buffer and transferred to 96-well white opaque flat bottom plates. Luciferase activity was measured using a Dual-Luciferase reporter assay system (Promega, Madison, Wisconsin, USA) and VICTOR Light (Perkin Elmer, Wallac, Turku, Finland).
In vivo antitumor study
Approximately 107 NCI-N87 and MKN45 cells in log growth phase were suspended in 0.2 ml phosphate buffered saline and subcutaneously injected into the flanks of severe combined immunodeficient (scid) mice. The animals were weighed weekly and tumour diameters measured twice weekly at right angles (dshort and dlong) with electronic calipers and converted to volume by the formula volume=[(dshort)2×(dlong)]/2. When the tumours reached volumes between 150 and 300 mm3, the mice were stratified into two groups of eight animals with approximately equal mean tumour volumes, followed by metformin treatment at 250 mg/kg orally daily for 25 days20 Control animals received vehicle (water) alone. Twenty-four hours after the final metformin administration, tumours were collected for western blot analysis and immunohistochemistry to assess the expression of HNF4α and WNT5A. When tumours reached ≥1500 mm3 or became necrotic, the animals were killed. All animal studies were approved by the National Cancer Center IRB Animal Committee.
Immunohistochemistry
Patient tumour samples
H&E-stained slides were reviewed and representative areas selected for tissue microarray. Normal mucosa and cancer tissues were selected, respectively. Cores 2 mm in diameter were taken from archival paraffin-embedded blocks using a trephine apparatus (Superbiochips Laboratories, Seoul, Republic of Korea). Tissue microarray blocks were sectioned at 3 μm thickness, dried for 1 h at 56°C, and immunohistochemical staining performed with the automated instrument BenchMark XT (Ventana Medical Systems, Tucson Arizona, USA) as follows: sections were deparaffinised and rehydrated with EZ Prep (Ventana) and washed with Tris-buffered saline. The antigens were retrieved by heat treatment in pH 8.0 Tris-EDTA buffer (Cell-Conditioning Solution, Ventana) at 95°C for 30 min for WNT5A. Endogenous peroxidases were blocked with 3% H2O2 for 10 min at room temperature. Blocking was performed with a ready-to-use protein blocker solution (Ventana) for 20 min at room temperature, followed by application of primary antibody at 42°C (WNT5A for 30 min at 1 : 5000 dilution (clone 6F2, Abgent, San Diego, California, USA, AO1264a), washing, incubation with HRP multimer-labelled secondary antibody (ultraView Universal DAB detection kit, Ventana) for 20 min at room temperature and staining using ultraView universal DAB kits (Ventana) for 8 min (with haematoxylin counterstain).
Mouse xenograft model
Immunohistochemical staining was performed on 4 μm tissue sections from paraffin embedded tissue blocks in the automated staining instrument Discovery XT (Ventana Medical Systems) using the Chromomap DAB Detection kit. For detection, anti-HIF-1α antibody (Abcam, Cambridge, UK), anti-cleaved caspase-3 (Cell Signaling Technology, Danverse, Massachusetts, USA), anti-Ki-67 (Abcam) and anti-CD31 (Abcam) were used. For grading, an H-score was determined for Ki-67, cleaved caspase-3 and HIF-1α.
Interpretation of immunohistochemistry
Patient tumour samples
We defined the intensity of WNT5A tissue staining as follows: (A) no signal or only a faint equivocal signal observed at 100× power was regarded as negative, 0; (B) >10% of tumour cells showing clear cytoplasmic signals at 100× power (similar to foveolar epithelial cells) was regarded weakly positive, +1; (C) if >10% of tumour cells showed obviously stronger signals with 40× power (similar to chief cells or the plasma cells for WNT5A), it was regarded as strongly positive, +2. We measured WNT5A positivity by the percentage of weakly or strongly positively staining tumour cells, scored as 0 (negative) if weakly or strongly positive cells were ≤50% and 1 (positive) if >50%. We used a log-rank test to examine the correlation between WNT5A levels and patient survival.
Mouse xenograft model
Immunohistochemical staining was graded as follows: intensities were graded (0=negative, 1=weak, 2=moderate, 3=strong) and the percentages of positive cells were taken to calculate the H-score, as follows: H-score=3×percentage of strongly staining cell(s)+2×percentage of strongly staining cell(s)+1×percentage of strongly staining cell(s), yielding a range of 0–300. For microvascular density (MVD) counting, microvesicles were stained using CD31 staining, with five fields chosen and counted at ×200.
Statistical analysis
For individual gene experiments, p values <0.05 were considered statistically significant, based on a Student t test or z-test, for comparisons of the experimental groups with control groups.
Results
Integrative transcriptomic analysis suggests HNF4α as a key signal mediator in GC
To comprehensively view the GC transcriptome in Caucasians, we performed whole-transcriptome profiling of 22 gastric tumours (and their matched normal tissues) using paired-end RNA-seq (Life Technologies SOLiD platform). Samples included stages I–III tumours from the distal, proximal, gastro-oesophageal junction and body regions of the stomach (see online supplementary table S1). On average, we obtained 53 million and 49 million properly paired short reads per tumour and normal tissue, respectively. Based on these mapped reads, we quantified the expression levels of 15 987 protein-coding genes.
To identify a robust gene signature of GC pathobiology across different populations, we integrated our Caucasian RNA-seq dataset with three other gene expression datasets previously collected from patients with GC20–22 (see online supplementary table S2). Across the four datasets, the expression levels of 12 587 genes were simultaneously measured, and we performed a differential expression analysis to identify those having significantly differential expression (see Materials and Methods). As shown in figure 1A, 396 genes were consistently identified as misexpressed by the four differential expression analyses. To identify genes showing the change direction (ie, upregulation or downregulation relative to those non-tumour samples) and the fold change, we classified them into five groups (figure 1B).

Differential expression analysis across multiple transcriptome-wide expression datasets. (A) Venn diagram of differentially expressed genes identified from the four expression datasets. (B) Classification of 396 genes into five groups: consistent upregulation with >2 fold; consistent upregulation with <2 fold; consistent downregulation with >2 fold; consistent downregulation with <2 fold; inconsistent changes. Differential expression of HNF4α in (C) Caucasian and (D) Asian RNA-seq datasets, respectively. Caucasian RNA-seq includes normal versus tumour stages I, II and III tissue samples. Asian RNA-seq includes normal versus tumour stages I, II, III and IV tissue samples.
To identify novel therapeutic targets we focused on the group that was consistently upregulated across the four analyses by >2 fold. Among the 148 genes, HNF4α appeared to be an interesting candidate. It is a key member of the nuclear receptor superfamily of ligand-dependent receptors and has been suggested to play a role in the development of colon, liver, pancreatic and lung cancers,8 ,25 ,26 and is highly expressed in intestinal type GC.27 Figure 1C and D show elevated expression of HNF4α in GC tumours from both Caucasian and Asian-Pacific patients. Given its robust expression and role as a transcriptional factor, we subsequently investigated HNF4α as a key regulator in the initiation and progression of GC.
HNF4α inhibition shows anti-tumour activity
To examine its neoplastic function, we performed shRNA-mediated HNF4α knockdown in NCI-N87 mouse xenografts. Using an effective shRNA (clone no 4, figure 2A), we found that the shRNA inhibition greatly reduced tumour growth compared with controls (figure 2B, C). As shown in figure 2D and supplementary figure S1A, HNF4α knockdown inhibited growth and angiogenesis, coincident with reduced HIF-1α. Further, we silenced HNF4α by siRNA in NCI-N87 GC cells. As shown in supplementary figure S1B, siRNA-mediated HNF4α knockdown also downregulated the cell cycle regulators cyclins A, B and D1. On days 2 and 3, respectively, we observed 62% and 68% silencing of HIF-1α, a regulator of cell cycle progression, glucose metabolism and angiogenesis.28 In addition, we performed shRNA-mediated HNF4α knockdown in MKN45 mouse xenografts and confirmed greatly reduced tumour growth compared with controls (see online supplementary figure S2A and S2B).

Hepatocyte nuclear factor-4α (HNF4α) inhibition shows antitumour activities. (A) A pool of five (short hairpin RNA) shRNA lentiviral particles targeting different regions of the HNF4α mRNA transcript and shRNA empty vectors was infected into NCI-N87 gastric cancer (GC) cells. Stably transfected cells were selected by puromycin, and clones 2 and 4 showed loss of HNF4α by immunoblotting. (B, C) Nine 5-week-old female BALB/C nude mice were randomly divided into two groups (4 shRNA control, 5 shRNA HNF4α). Approximately 107 shRNA control (CTRL) and HNF4α (#2 and #4) shRNA-infected NCI-N87 cells in 100 µL phosphate-buffered saline were inoculated subcutaneously. All animals were sacrificed on day 31 after injection. ***p<0.001. (D) Immunoblotting shows antiproliferative activity with loss of hypoxia-inducible factor 1α (HIF-1α) from HNF4α shRNA-transduced xenograft model tumours.
To further evaluate HNF4α as a therapeutic target, we showed the HNF4α antagonist BI6015 to variably inhibit HNF4α expression in a panel of GC cell lines (figure 3A) while effectively reducing GC cell survival (EC50 values ranging from 964 nM to 4.3 μM, figure 3B). Therefore, pharmacological inhibition elicited the same effects as transcript-specific targeting (shRNA), which suggests the drug was specific for HNF4α and not non-specifically toxic (figure 3C). These results strongly suggest that HNF4α is a key GC signal mediator, representing a promising therapeutic target.

Effect of the hepatocyte nuclear factor-4α (HNF4α) antagonist BI6015 on gastric cancer (GC) cell lines. (A) Expression of HNF4α in GC cell lines treated with or without BI6015 using quantitative RT-PCR (*p<0.05) as well as the molecular structure of BI6015. (B) EC50 growth inhibition values of BI6015 in various GC cell lines. (C) BI6015 shows different levels of growth inhibition on five GC cell lines.
HNF4α is downstream of AMPKα and is modulated by the AMPK agonist metformin
We next examined how HNF4α is regulated in GC by specifically focusing on AMPKα2, a key member of the AMPK family that maintains energy balance and is downregulated in primary breast and ovarian cancers.29 We recently reported that AMPKα2 is a potential tumour suppressor in Asian-Pacific patients with GC, having clear mRNA loss in early stage tumours compared with normal tissues or advanced tumours.20 Importantly, HNF4α has been suggested as a downstream target of AMPKα signalling.30 ,31 For example, Ribeiro et al showed that AMPK directly phosphorylates HNF4α Ser304 in its ligand-binding domain, inhibiting dimerisation and DNA binding.31 ,32
Based on an independent set of gastric tumours (n=10 samples per stage group), we confirmed loss of AMPKα2 expression in early stage tumours (t test, p=0.025, see online supplementary figure S3A). Through RNA-seq analysis across different tumour stages we found that, in both Caucasian and Asian-Pacific patients, HNF4α and AMPKα2 showed opposite mRNA expression patterns: such anticorrelation (upregulation of HNF4α and loss of AMPKα2) was stronger in early stage tumours than in late stage tumours (figure 1C, D, see online supplementary figures S3B and S3C). Given this negative correlation of AMPKα2 and HNF4α expression, we examined a possible mechanistic connection between the two genes. First, we confirmed AMPKα2 protein expression in a panel of GC cell lines and xenograft tissues (see online supplementary figures S4A and SB, respectively) and, second, we found that HNF4α protein expression varied among GC cell lines (see online supplementary figure S4C).
To examine the interaction between AMPKα2 and HNF4α, we used metformin (an AMPK activator) to treat four GC cell lines (NCI-N87, AGS, HS746T and MKN45). As expected, metformin activated the tumour suppressing liver kinase B1 (LKB1), an upstream regulator of AMPK,33 in these cell lines (see online supplementary figure S5). Metformin treatment also increased AMPKα2 expression levels (figure 4A) and decreased HNF4α expression (figure 4B) in GC cells, suggesting that AMPK activation may downregulate HNF4α. To investigate possible anticancer effects of metformin-mediated HNF4α inhibition, we assessed the viability of 10 GC cell lines after metformin treatment, including the four abovementioned cells and another six cell lines (SNU-1, SNU-16 and SNU-620; and three human GC cell lines newly established from Korean patient primary tumours, NCC-19, NCC-59 and SNU-1967). We observed strong growth inhibition in all 10 GC cell lines (figure 4C) following perturbation of AMPKα-HNF4α signalling by metformin. The levels of cleaved caspase 3 (c-caspase 3) and poly ADP-ribose polymerase (cPARP) decreased upon metformin treatment, indicating that apoptosis is not the only cause of growth inhibition (see online supplementary figure S6). In our GC panel cell lines we observed G2/M arrest and suppression of cyclins A, B and D1 (figure 4D and E), and additionally detected increased sub-G0 populations (see online supplementary figure S7). Next, we performed apoptosis and necrosis analysis using flow cytometry and detected various apoptotic effects in a few GC cell lines (see online supplementary figure S8). Moreover, no autophagy was detected upon metformin treatment (data not shown). Overall, these results indicate that HNF4α is downstream of AMPKα, and the antiproliferation effect of metformin is largely due to HNF4α inhibition-induced cell cycle arrest, although we did see some induction of cell death.
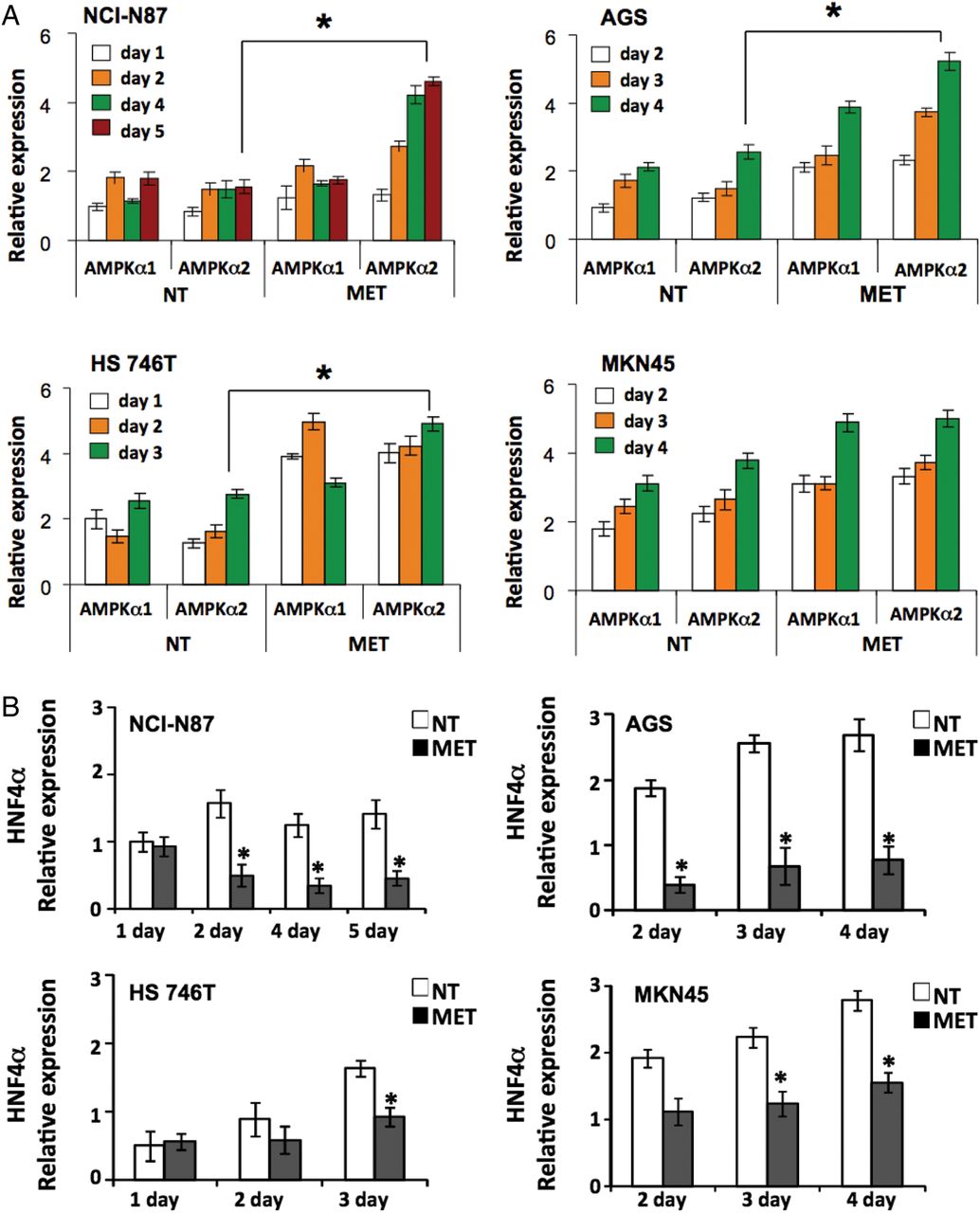
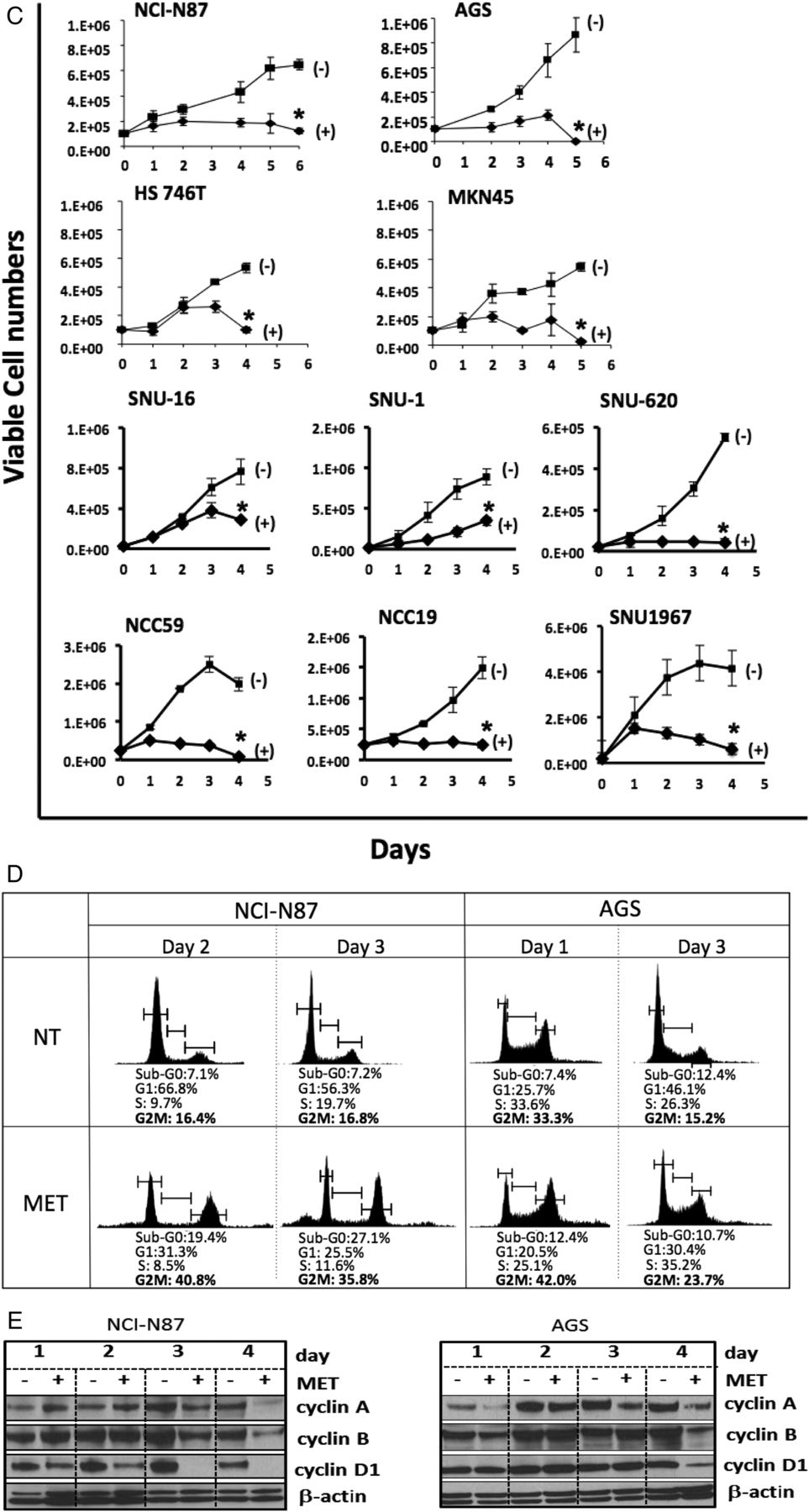
AMP-activated protein kinase (AMPKα) activation and hepatocyte nuclear factor-4α (HNF4α) inhibition by metformin show antiproliferation activity in gastric cancer (GC) cell lines. (A) Expression of AMPKα1 and AMPKα2 in four GC cell lines (NCI-N87, AGS, HS 746T and MKN45), with and without metformin treatment, as measured by quantitative RT-PCR (*p<0.05). For NCI-N87, white bars=day 1, orange bars=day 2, green bars=day 4, red bars=day 5; for AGS and MKN45, white bars=day 2, orange bars=day 3, green bars=day 4; and for HS 746T, white bars=day 1, orange bars=day 2, green bars=day 3. (B) Expression of HNF4α in four GC cell lines, with and without metformin treatment (white bars=non-treated (NT), black bars=metformin-treated (MET)), as measured by quantitative RT-PCR (*p<0.05). (C) Growth inhibition observed in 10 GC cell lines (NCI-N87, AGS, HS 746T, MKN45, SNU-1, SNU-16, SNU-620, NCC59, NCC19 and SNU1967) following metformin treatment (squares=non-treated (NT), diamonds=metformin-treated (MET)). Cells were counted from the onset of metformin treatment (day 0) (*p<0.05). (D) Metformin mechanism of action through AMPKα activation and HNF4α inhibition, resulting in G2/M arrest and antiproliferation of GC cell lines. Two GC cell lines (NCI-N87 and AGS) were treated with metformin and cells were fixed and analysed for DNA content using propidium iodide and flow cytometry. Metformin-treated cells were compared with non-treated cells. The percentage of cells in each cell cycle stage was calculated. Cell cycle arrest in the G2M phase was observed following metformin treatment. (E) Immunoblotting shows the expression levels of cyclins A, B and D1. β-actin was used as control. Decreased cyclin D1 levels were apparent in both NCI-N87 and AGS cell lines upon metformin treatment.
WNT5A is a target gene of HNF4α with potential prognostic value
We recently reported WNT5A as a direct target gene of HNF4α in GC.34 For further confirmation, we silenced HNF4α using siRNA in six GC cell lines (NCI-N87, MKN45, NCC19, NCC24, NCC59 and SNU1967) and performed TCF/LEF reporter luciferase assays to monitor WNT signalling. In concert with silencing of HNF4α (figure 5A), five of the six GC cell lines showed significant suppression of TCF/LEF (figure 5B) and three GC cell lines (MKN45, NCC24 and NCC59) showed downregulation of WNT5A (figure 5C) under HNF4α shRNA knockdown.

Effects of hepatocyte nuclear factor-4α (HNF4α) knockdown on WNT5A in gastric cancer (GC). (A) Monitoring of HNF4α activity through HNF4α reporter luciferase assay under siRNA-HNF4α in six GC cell lines (diffuse type: SNU1967 and NCC24; intestinal type: NCC19 and NCC59) (*p<0.05). (B) Monitoring of WNT signal pathway activity through TCF/LEF reporter luciferase assays following siRNA-HNF4α knockdown in six GC cell lines (*p<0.05). (C) Suppression of WNT5A mRNA expression levels by negative control (white bars), HNF4α shRNA (black bars) or WNT5A (line filled bars) in three GC cell lines.
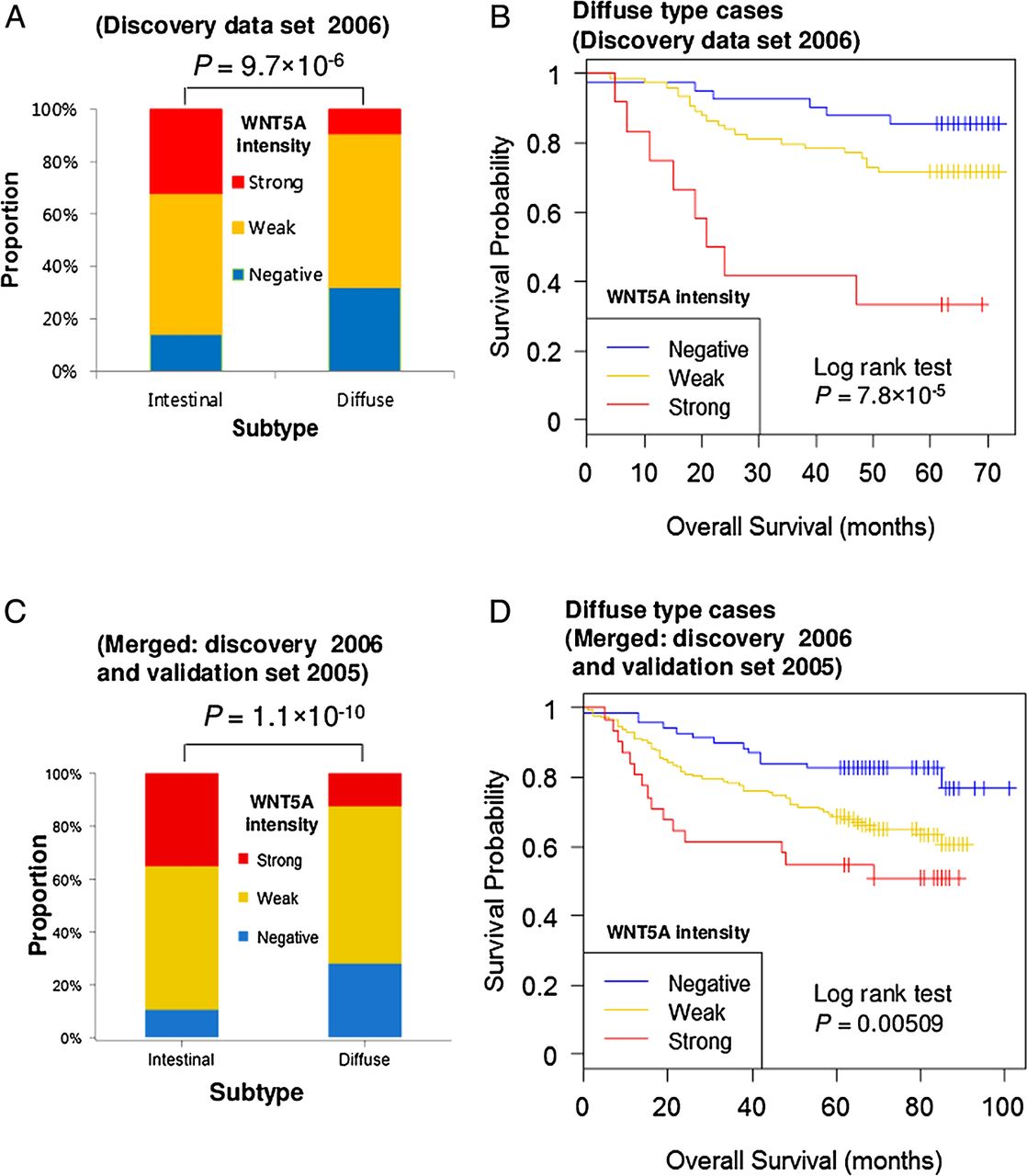
To explore the clinical relevance of WNT5A, we performed immunohistochemical staining in two cohort (251 discovery set and 251 validation set) cases of advanced GC obtained from Asian-Pacific patients (clinical characteristics summarised in online supplementary table S3). Figure 6A shows representative results of WNT5A immunostaining of GC tumours of the merged two-cohort study. In non-neoplastic tissues, the neck area of gastric glands and whole length of intestinal metaplastic glands were positive for WNT5A. Overall, WNT5A expression was observed in 195 (77.7%) and 214 (85.3%) samples in the discovery and validation sets, respectively. High expression (intensity 2+) was observed in 57 (22.7%) and 68 (27.1%) samples. The relationships between WNT5A intensity and various clinical variables are provided in online supplementary table S3. Interestingly, WNT5A showed high expression levels in Lauren intestinal type GC (figure 6A, B, ANOVA test, p=9.7×10−6), while negative WNT5A staining levels correlated with diffuse type GC, a relationship maintained in both the validation and merged sets (figure 6C, D and see online supplementary table S4). In the discovery case set (2006), higher expression of WNT5A related to older age and more advanced cancer status (deeper invasion and increased nodal metastasis); however, these relationships were not present in the validation set (see online supplementary table S4 and S5 and supplementary figure S9). These inconsistencies could be caused by the variability of the immunohistochemical stains or the differences between the two cohorts.

WNT5A is a potential prognostic marker in gastric cancer (GC) of the diffuse type. (A) WNT5A shows a strong bias with regard to the Lauren types in discovery data set (2006). (B) Kaplan–Meier plot showing correlation of WNT5A intensity with patient overall survival in the diffuse type in the discovery dataset (2006). (C, D) Merged (discovery data set 2006 and validation data set 2005) dataset shows WNT5A as a prognostic marker of diffuse type GC.
WNT5A expression levels showed no prognostic significance when analysed for all the cases (see online supplementary figures S10A and C) without regard to Lauren type. However, when the cases were separated into Lauren types, strong intensity of WNT5A was associated with worse overall survival in the diffuse GC type (log rank test p=7.8×10−5, figure 6B) of the discovery cohort. While we could not show this association repeatedly in the validation cohort (see online supplementary figure S11A and supplementary table S5), the association with worse overall survival was still present in the merged set (figure 6D). No significant association with WNT5A levels was found with the intestinal GC type in two cohorts (see online supplementary figures S10B and D). These results suggest the potential of WNT5A as a molecular marker to facilitate patient stratification. The higher expression of WNT5A in intestinal type cancer and non-neoplastic glands of intestinal metaplasia may be a phenomenon that accompanies intestinal differentiation. In diffuse type cancers, however, high expression of WNT5A is not a phenomenon accompanied by differentiation and may represent a functional acquisition to more aggressive behaviour.
Animal models confirm the AMPK-HNF4α-WNT5A signalling cascade
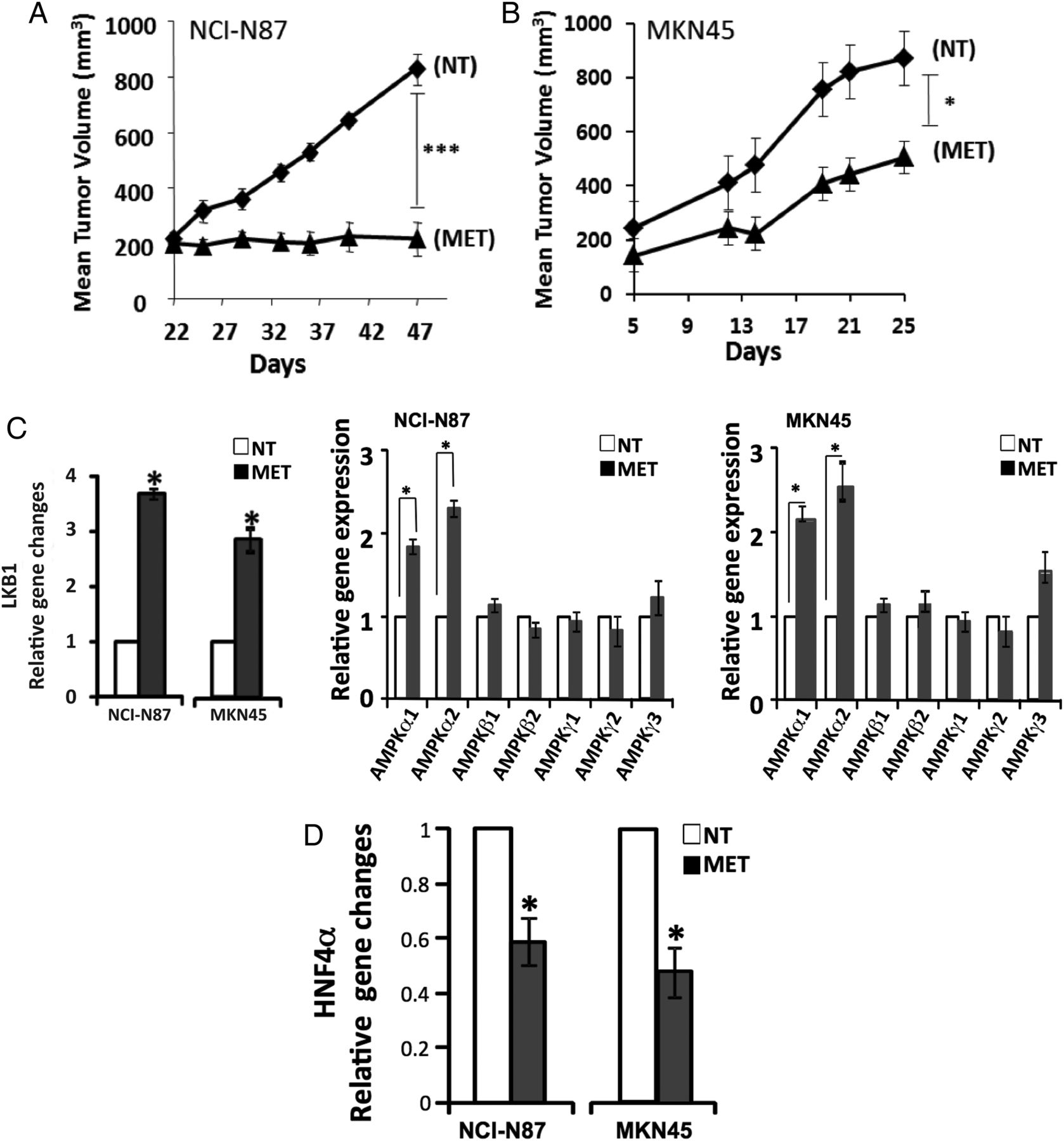
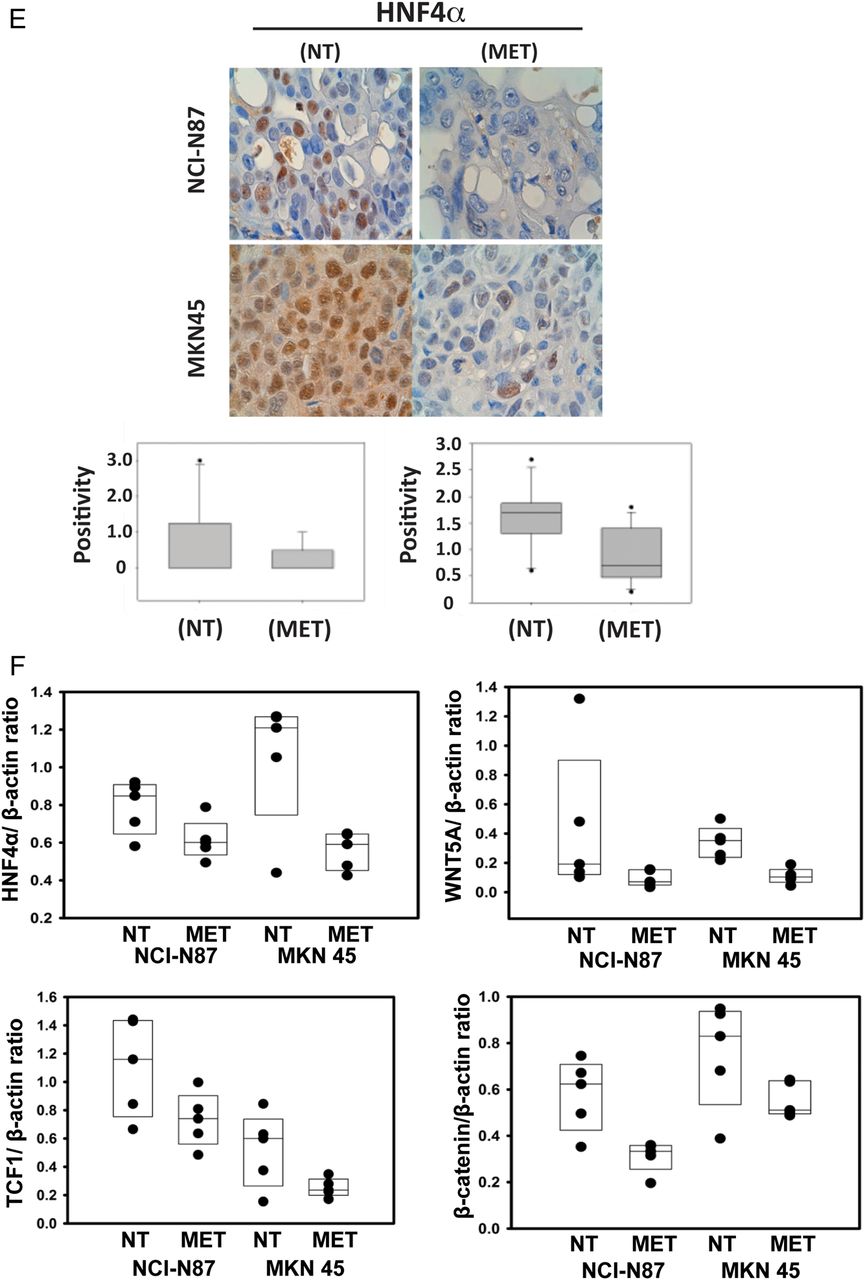
From our previous study and the above GC cell line perturbation experiments (metformin treatment and siRNA-mediated knockdown), we elucidated the context of HNF4α signalling in GC, with upstream regulators LKB1-AMPK and downstream targets WNT5A–HIF-1α. To confirm the above finding in a more biologically relevant context, we performed experiments in two xenograft (NCI-N87 and MKN45) mouse models, showing metformin to have strong antitumor activity (figure 7A, B). Importantly, we found the regulatory relationships to be consistent with our observations in the cell line studies. Upon metformin treatment, LKB1 expression increased 3.6-fold in NCI-N87 cells and 2.8-fold in MKN45 cells compared with no treatment; AMPKα expression also significantly increased (figure 7C). Concomitantly, HNF4α mRNA levels decreased by 44% in NCI-N87 cells and 50% in MKN45 cells compared with no treatment (figure 7D). Immunohistochemistry also revealed HNF4α protein downregulation in xenograft models (figure 7E), and WNT5A and its direct downstream target genes, β-catenin and TCF1, were all downregulated by metformin (figure 7F). These in vivo results further validate our immunohistochemistry and RNA-seq findings, showing that LKB1–AMPK–HNF4α–WNT5A–HIF-1α represents a key signal cascade in GC tumorigenesis. These results further validate our computational approach (‘PATHOME’), in which we implicated an HNF4α-WNT5A signal coupling in GC,34 an approach we believe improves on the mere examination of gene ontology or gene set enrichment analysis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Animal models confirm the liver kinase B1/AMP-activated protein kinase/hepatocyte nuclear factor-4α (LKB1/AMPK/HNF4α) signalling cascade in gastric cancer (GC) mouse models. (A, B) Antitumor activity of metformin treatment on NCI-N87 and MKN45 GC mouse xenograft models (scid mice, n=8 per cell type). Cells were subcutaneously injected and grown for 22 days for NCI-N87 cells and 5 days for MKN45 cells before treating with metformin. Tumour size growth was measured and compared between mice treated with or without metformin, as depicted by the growth curve (diamond=non-treated (NT); triangle=metformin-treated (MET)) (*p<0.05, ***p<0.001). (C) AMPK subunit expression levels with and without metformin after 25 days (open bars=NT, black bars=MET). The expression levels of AMPKα1 and AMPKα2 were significantly higher when treated with metformin, but not those of AMPKβ1, AMPKβ2, AMPKγ1, AMPKγ2, AMPKγ3 (*p<0.05). (D) Expression level of HNF4α with and without metformin at 25 days (open bars=NT, black bars=MET). HNF4α expression level decreased upon metformin treatment. *p<0.05. (E) Immunohistochemistry results showing WNT5A staining of mouse model xenograft tumours. WNT5A levels in tumours strongly decreased when mice were treated with metformin. The stain was scored as 0–3, representing weakly or strongly positively stained cells. (F) Xenograft model protein levels for β-catenin and TCF1, the downstream genes of the HNF4α, WNT5A and WNT pathway. Mice were untreated or treated with metformin for 25 days. Four graphs represent quantified immunoblots using ImageQuant software normalised to β-actin.
Discussion
In this study we profiled GC transcriptomes from Caucasian as well as Asian-Pacific patients, providing a critical foundation for the molecular basis of this disease across different ethnic groups. Importantly, instead of selecting candidate genes from a single dataset, we undertook a sub-pathway approach, integrating multiple expression datasets for several different patient populations (Asian-Pacific and Caucasian).34 Given the high biological heterogeneity of GC, this integrative process allowed us to define a small set of genes with the most robust expression signatures that correlate with GC tumorigenesis.
Those analyses identified HNF4α as a key GC oncogene. HNF4α is a nuclear receptor that activates the expression of genes involved in endoderm development and glucose, fatty acid and cholesterol metabolism, and its isoforms have been studied in the liver.35 ,36 Previous studies suggested HNF4α as a potential direct or indirect target for pharmacological drugs that act on the intestinal epithelium (a primary site of metabolic control),32 and one HNF4α isoform was shown to correlate with Epstein–Barr viral infection in GC tumours.37 To assess the function of HNF4α, we performed GC cell perturbation experiments through shRNA or direct antagonists or via metformin, a widely used antidiabetes drug.38 In agreement with our results, metformin was previously found to inhibit MKN-1, -45 and -74 GC cell lines and xenograft growth through its effects on cell cycle regulatory proteins.39 Here we further show that metformin treatment activated LKB1-AMPK activation while downregulating HNF4α. Our study demonstrated that indirect (AMPK) or direct HNF4α inhibition robustly downregulated HIF-1α, which is consistent with a proposed role for AMPK in suppressing the ‘Warburg effect’ anaerobic metabolism characteristic of the malignant phenotype.40
Importantly, our results show that HNF4α antagonists have antitumor activity and also that HNF4α expression can be modulated by metformin through AMPK signalling, providing insight into possible GC drug combinations. Moreover, HNF4α can regulate WNT signalling through its direct target WNT5A that is a potential prognostic marker, and our finding of WNT5A suppression by AMPK agrees with a previous study showing upregulation of hepatic WNT5A under fasting conditions.41 Together, our results highlight the potential of the AMPKα–HNF4α–WNT5A signalling cascade as a promising targetable pathway for future drug development.
In summary, we believe that our approach of identifying subsets of signal pathways that associate with specific hallmarks of cancer represents a significant improvement over previous methods considering only specific gene ontologies or gene set enrichment. That specific algorithm, designated ‘PATHOME’,34 previously postulated the existence of an HNF4α/WNT5A signal coupling, a computational result that we have experimentally validated in the current work. Together, our results highlight the potential of the AMPKα–HNF4α–WNT5A signalling cascade as a promising targetable pathway for future GC drug development.
Acknowledgments
We thank LeeAnn Chastain for editorial assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
HRC, SN, and M-CK are equally contributed first authors. TP, HL, and YHK are equally contributed corresponding authors.
Contributors TP, HL and YHK conceived and designed the experiments. DH, SN, IH, HL and YHK analysed the data. HRC, K-TK, M-CK, HRJ, HSP, YG, GP and YHK performed the experiments. HL, TP and YHK wrote the paper. All authors have read and approved the manuscript for publication.
Funding This study was supported by grants from the National Cancer Center of the Republic of Korea, NCC-1210350-2 (to YHK), NCC-1210460 (to SN); the National Research Foundation of Korea (NRF) grant funded by the Republic of Korea (MSIP) 2012R1A3A2026438, 2008–0062618, 2013M3A9C4078158 (to TP); grants from the U.S. National Institutes of Health, CA95060, CA129616, CA17094 and CA98920 (to GP), CA143883 and CA016672 (to HL); UTMDACC-G.S. Hogan Gastrointestinal Research Fund and the Lorraine Dell Program in Bioinformatics for Personalization of Cancer Medicine (to HL).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The gene expression data in this study can be found online at the Gene Expression Omnibus (GSE63288).