Histone Deacetylase Inhibitors as Anticancer Drugs


1
Department of Pediatric Hematology and Oncology, 2nd Faculty of Medicine, Charles University and University Hospital Motol, V Uvalu 84/1, Prague 5 CZ-150 06, Czech Republic
2
Department of Biochemistry, Faculty of Science, Charles University, Albertov 2030/8, Prague 2 CZ-128 43, Czech Republic
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(7), 1414; https://doi.org/10.3390/ijms18071414
Submission received: 14 May 2017
/
Revised: 11 June 2017
/
Accepted: 27 June 2017
/
Published: 1 July 2017
(This article belongs to the Special Issue Cancer Epigenetics)
Abstract
:Carcinogenesis cannot be explained only by genetic alterations, but also involves epigenetic processes. Modification of histones by acetylation plays a key role in epigenetic regulation of gene expression and is controlled by the balance between histone deacetylases (HDAC) and histone acetyltransferases (HAT). HDAC inhibitors induce cancer cell cycle arrest, differentiation and cell death, reduce angiogenesis and modulate immune response. Mechanisms of anticancer effects of HDAC inhibitors are not uniform; they may be different and depend on the cancer type, HDAC inhibitors, doses, etc. HDAC inhibitors seem to be promising anti-cancer drugs particularly in the combination with other anti-cancer drugs and/or radiotherapy. HDAC inhibitors vorinostat, romidepsin and belinostat have been approved for some T-cell lymphoma and panobinostat for multiple myeloma. Other HDAC inhibitors are in clinical trials for the treatment of hematological and solid malignancies. The results of such studies are promising but further larger studies are needed. Because of the reversibility of epigenetic changes during cancer development, the potency of epigenetic therapies seems to be of great importance. Here, we summarize the data on different classes of HDAC inhibitors, mechanisms of their actions and discuss novel results of preclinical and clinical studies, including the combination with other therapeutic modalities.
1. Introduction
Cancer chemotherapy has been one of the major medical advances in the last few decades. However, the drugs used for this therapy have a narrow therapeutic index, and the produced responses are often only palliative as well as unpredictable. Such approaches, although directed toward certain biomacromolecules, do not discriminate between rapidly dividing non-malignant and cancer cells. In contrast, targeted therapy that has been introduced in recent years is directed against cancer-specific targets and signaling pathways, and thus has more limited nonspecific mechanisms.
A crucial role for epigenetic mechanisms in cancer development is demonstrated by a number of studies. Carcinogenesis cannot be explained only by genetic alterations, but also involve epigenetic processes (DNA methylation, histone modifications and non-coding RNA deregulation). Histone modifications include H3 and H4 histones lysine deacetylation that leads to chromatin decondensation [1]. These alterations influence gene transcription including upregulation of several anti-oncogenes and DNA repair genes [2]. Thus, the epigenetic processes have emerged as novel therapeutic targets in numerous investigations.
The importance of histone deacetylase (HDAC) enzymes in organisms has been demonstrated using the studies with mice knocked out on members of class I HDACs. HDAC1-null mice die prenatally with severe proliferation defects and general growth retardation; HDAC2-null mice die the first day after birth for cardiac malformations; and HDAC3-null mice die prenatally for defects in gastrulation [3,4]. HDACs seem to be important for gene expression [5]. It has been described several times that their levels vary greatly in cancer cells and differ according to the tumor type. HDAC1 is highly expressed in prostate, gastric, lung, esophageal, colon and breast cancers [6,7,8]. High levels of HDAC2 were found in colorectal, cervical and gastric cancers [9,10]. In addition, HDAC3 is overexpressed in colon and breast tumors [11], whereas HDAC6 is highly expressed in mammary tumors, HDAC8 is overexpressed in neuroblastoma cells and HDAC11 mainly in rhabdomyosarcoma [12,13,14]. Increased expression of different HDAC and/or histone hyperacetylation in different cancers is caused by different mechanisms which may affect effects of individual HDAC inhibitors.
The in vitro analysis of different HDACs isoforms using siRNA (small interfering RNA) against HDAC1, HDAC2 and HDAC3 on several ovarian carcinoma cells (SKOV3, OVCAR3, IGROV-1, ES-2, TOV112D, A2780 and A2780/CDDP) showed that knockdown of HDAC1 inhibits proliferation and tumorigenicity, while knockdown of HDAC3 reduces cell migration with an increase in E-cadherin. On the contrary, knockdown of HDAC2 has no effect on proliferation and tumorigenicity, or cell migration [15].
In this review, we describe different classes of HDAC inhibitors and mechanisms of their actions, and discuss novel data found in several preclinical experiments and clinical studies. Moreover, we discuss their combination with other therapeutic modalities, particularly with DNA-damaging compounds.
2. Acetylation and Deacetylation
Modification of histones by acetylation plays a key role in epigenetic regulation of gene expression by changing the structure of chromatin and by modulating the accessibility of transcription factors to their target DNA sequences [16]. Histone acetylation is enriched in transcriptionally active regions of the genome, especially at proximal promoters and enhancers, and facilitates the binding of transcription factors. Acetylation loosens contact between core nucleosome proteins and DNA, thereby making transcription factor binding sites more accessible, recruitment of bromodomain proteins which recognize acetylated lysine residues on the N-terminal tails of histones and are actively recruited to enhancer and promoter regions [17]. The acetylation state of histones and other proteins is maintained by histone acetyltransferases (HAT) and HDAC. HATs catalyze the transfer of an acetyl group from acetyl-CoA to the ε-NH2 group of lysine residues in proteins, while HDACs remove it [12]. Acetylation status of non-histone proteins modifies many cellular functions e.g., mRNA splicing, transport and integrity; translation; activity, localization, stability and protein interactions [18]. Of the proteins whose functions are modulated by HDACs, the p53 (tumor protein 53), RUNX3 (Core binding factor α3 subunit), STAT3 (Signal transduction and activation of transcription 3), β-catenin, estrogen receptor, Myc (Avian myelocytomatosis viral oncogene homolog), EKLF (Erythroid Kruppel-like factor), GATA family (GATA-binding factors), HIF-1α (Hypoxia-inducible factor 1α), MyoD (Myogenic regulatory factor), NF-κB (Nuclear factor κB) or Foxp3 (Forkhead box P3 protein), are the most important in cancer (for review, see [19]).
Based on the homology to their yeast analogs, HDACs are divided into four classes. Class I, located in the nucleus, includes HDACs 1, 2, 3 and 8. HDACs 4, 5, 7 and 9 are members of class IIa, and class IIb (isoforms 6 and 10) are located in both cytoplasm and nucleus. Class IV (only the isoform HDAC11) exhibits some features of classes I and II. NAD+-dependent homologs 1–7 of the yeast Sir2 proteins (sirtuins) are designed as the class III of HDACs [20]. To remove the acetyl groups of proteins, the HDACs utilize two different mechanisms. These mechanisms act as the base for their classification into two distinct families. The “classical family” comprises of Zn2+-dependent HDACs (classes I, II and IV). The Zn2+ ion stabilizes the acetylated substrate in the catalytic center of the enzyme and polarizes the carbonyl group making the carbon to be a better target for nucleophilic water molecules [21]. The further HDAC family is NAD+-dependent, being capable of forming O-acetyl ADP ribose and nicotinamide as a result of the acetyl transfer [22].
Most of the classic HDACs form large complexes with multiple transcriptional co-repressors (such as Sin3 (Transcriptional regulatory protein SIN3), NuRD (Nucleosome remodeling and deacetylase complex), NcoR (Nuclear receptor co-repressor) and SMRT (Silencing mediator of retinoic acid and thyroid hormone receptors) with chromatin remodeling activity [23]. HDAC complexes can contribute to gene repression through two mechanisms—specific targeting by repressors and the constitutive association with chromatin [24,25].
HATs, which have the counterbalancing effect of HDAC, are a diverse set of multi-subunit complexes. They are divided into Gcn5 N-acetyltransferases (GNATs) and MYST HATs. GNATs include Gcn5 (General control non-repressed protein 5), PCAF (p300-CREB-binding protein-associated factor), Elp3 (Elongator protein 3), Hat1 (Histone acetyltransferase 1), Hpa2 (Hrp-associated 2 protein) and Nut1 (Component of the RNA polymerase II mediator complex). MYST is an acronym for the members of a protein family: Morf, Ybf2 (Sas3), Sas2 and Tip60. Although these two groups of HATs are the main ones, other proteins such as p300/CBP (CREB-binding protein), Taf1 (TATA box-binding protein associated factor 1) and a number of nuclear receptor coactivators show intrinsic acetylase activity. However, they do not contain true consensus HAT domains and therefore represent an “orphan class” of HAT enzymes [26].
3. HDAC Inhibition and Its Effects
Histone acetylation has been shown to be an important regulatory mechanism that controls transcription of approximately 2–10% of genes [27]. Deacetylation of histones causes chromatin condensation, while decondensation is caused by increased acetylation [1]. Such changes might result in decreased or increased gene transcription. However, there are other proteins than histones (e.g., chromatin remodeling proteins, DNA-binding nuclear receptors, DNA repair enzymes, signaling mediators, structural proteins, transcription coregulators, DNA-binding transcription factors) whose activity is affected by acetylation. In particular, the function of transcription factors can be positively or negatively affected. It provides an explanation why gene expression determined by HDAC inhibition, is not always increased even if the chromatin structure is loosened. The changes in HAT/HDAC activity balance can: (I) lead to an altered gene expression profile as well as to the change of some signaling pathways e.g., ERK (Extracellular signal-regulated kinase) and Wnt (Wingless/Int-1) pathways; (II) affect proteasomal degradation; (III) influence protein kinase C activity and (IV) change DNA methylation status. In various cancer cells, the shift to an increased acetylation/deacetylation ratio by HDAC inhibition was found to have a substantial effect on their fate (for overview see [19]).
HDAC inhibitors may act specifically against the only several types of HDACs (HDAC isoform-selective inhibitors), but also against all types of HDACs (pan-inhibitors). HDAC inhibitors can be classified as members of five classes of compounds: (I) hydroxamic acids (hydroxamates); (II) short chain fatty (aliphatic) acids; (III) benzamides; (IV) cyclic tetrapeptides; and (V) sirtuin inhibitors including the pan-inhibitor nicotinamide and the specific SIRT1 and SIRT2 inhibitors sirtinol and cambinol, respectively (for overview, see [28] and Table 1).
(I) Whereas trichostatin A (TSA) is an HDAC inhibitor used only in laboratory experiments because of its toxicity, vorinostat (suberoylanilide hydroxamic acid, SAHA) which has been already approved by United States Food and Drug Administration (FDA) as the first HDAC inhibitor, is utilized for the treatment of relapsed and refractory cutaneous T-cell lymphoma (CTCL). Belinostat (PXD-101) (approved for therapy of peripheral T cell lymphoma (PTCL)), panobinostat (LBH589) (approved for therapy of multiple myeloma) and the compounds recently tested in clinical studies, i.e., givinostat (ITF2357), resminostat (4SC201), abexinostat (PCI24781) and quisinostat (JNJ-26481585), are the pan-HDAC inhibitors. Selective HDAC inhibitors in the group of hydroxamic acids, including rocilinostat (ACY1215), which is selective inhibitor of HDAC class II, as well as practinostat (SB939) that inhibits all classical classes of HDACs (I, II and IV) and CHR-3996, the selective inhibitor of class I, are under clinical studies.
(II) The short chain fatty acids, valproic acid (VPA), butyric acid and phenylbutyric acid, are known to be weak inhibitors of HDAC class I and IIa and I and II, respectively. VPA is registered for the therapy of epilepsy, bipolar disorders and migraines, and is now together with other short chain fatty acids HDAC inhibitors tested in clinical studies as anticancer drugs.
(III) Among the benzamides that are now tested in clinical studies, entinostat (MS-275-SNDX-275), tacedinaline (CI994) and 4SC202 inhibit the class I HDACs. Mocetinostat (MGCD0103) is a selective inhibitor of classes I and IV HDACs.
(IV) The cyclic tetrapeptides include the bicyclic depsipeptide romidepsin (FK228, FR901228), a compound that has been approved by FDA and EMA (European Medicines Agency) to treat CTCL. It is a prodrug, which is reductively activated to a metabolite containing a thiol group that chelates the zinc ions in the active center of the HDAC of class I [29].
(V) Sirtuin inhibitors include the pan-inhibitor nicotinamide and the specific SIRT1 and SIRT2 inhibitors sirtinol, cambinol and EX-527. They might act against different types of neurodegenerations and cancers [30].
4. Mechanisms of HDAC Inhibitors Action
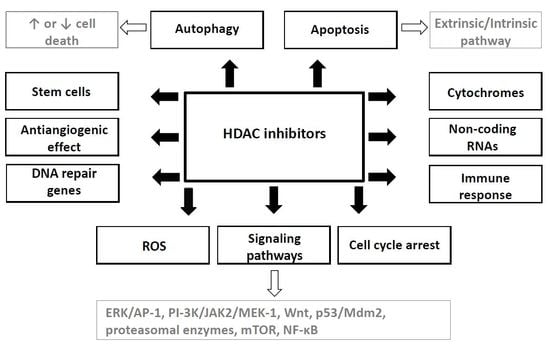
HDAC inhibitors induce cancer cell cycle arrest, differentiation and cell death. Moreover, they reduce angiogenesis and modulate immune response. Hypothesis of “epigenetic vulnerability of cancer cells”, which has been proposed by Dawson and Kouzarides [38], is a cause of relative specificity of HDAC inhibitors. This hypothesis supposed that normal cells have in contrast to some cancer cells multiplied epigenetic regulatory mechanisms. Therefore, HDACs may be essential for the maintenance of a set of key genes required for survival and growth of cancer cells but not of normal ones [38].
Mechanisms of anticancer effects of HDAC inhibitors are not uniform; they may be different and depend on a type of cancer, on the individual HDAC inhibitor and its dose as well as on some other factors [39]. For example, VPA has shown to inhibit the invasiveness in bladder cancer but not in prostate cancer cells [40], while it has not induced cell cycle inhibition in some neuroblastoma cell lines such as SH-SY5Y and SK-N-BE [41].
4.1. Cell Cycle Arrest
Cell cycle arrest induced by HDAC inhibitors is caused by several mechanisms; the most important seems to be the increased expression of cell cycle genes such as CDKN1A (Cyclin dependent kinase inhibitor p21) as described in a variety of cancer cells [42,43,44]. Its product blocks the formation of dimers from cyclins and cyclin dependent kinases (CDKs) that induce cell cycle arrest and inhibition of cell differentiation [43,44]. The expression of p21 is modulated by protein p53 that interacts with p21 promoter, competing with HDAC1, which decreases transcription of p21 [45]. After treatment with HDAC inhibitors, the HDAC1 protein is released from the Sp1 (Promoter-specific RNA polymerase II transcription factor), which increase p21 expression. Furthermore, HDAC inhibition increases acetylation of the p53 protein which results in an increase in its half-life [46], thereby improving the interaction with the p21 promoter [47]. Moreover, the p53 protein interactions with its activators ASPPs (Ankyrin-repeat-, SH3-domain- and proline-rich region containing proteins), 53BP1 (p53-binding protein), TiP60/hMOF (Human males absent on the first), hCAS/CSE1L (Cellular apoptosis susceptibility protein), and HZF (Hematopoietic zinc finger) are regulated by its acetylation status which is influenced by HDAC inhibitors [48]. Finally, the p21 levels are increased, thereby mediating cell cycle arrest and apoptosis [43,49,50]. HDAC inhibitors can also inhibit expression of genes coding cyclin D and cyclin A resulting in the absence of activities of the corresponding kinases, CDK2 and CDK4 [44,51]. In addition, the HDAC inhibitors may increase the stability and transcriptional activities of RUNX3, which mediates induction of p21 and product of anti-apoptotic gene Bim (Bcl-2-interacting mediator of cell death) [52,53,54,55].
4.2 Apoptosis Induction
HDAC inhibitors induce apoptosis in tumor cells by regulation of pro-apoptotic and anti-apoptotic genes (for a review see [56,57,58]). The mechanisms by which different HDAC inhibitors induce apoptosis include activation of both extrinsic and intrinsic apoptotic pathways.
Initiation of the extrinsic apoptotic pathway by HDAC inhibitors was proven in many in vitro experiments. HDAC inhibitors have been demonstrated to influence death receptors TRAIL (TNF related apoptosis inducing ligand), DR5 (Death receptor 5), Fas (TNF superfamily 6), TNF (Tumor necrosis factor) and TNF-related ligands Fas-L, LIGHT (TNF superfamily member 14) and TLA1 (Transparent leaf area peptide). Inhibition of those death receptors and their ligands inhibits apoptosis induced by HDAC inhibitors [57,59,60,61]. In vivo experiments with xenograft using tumor cells with TRAIL and Fas suppressed by siRNA showed a significant decrease in apoptosis after treatment with VPA [62]. HDAC inhibitors also activate intrinsic apoptotic pathway. They regulate transcription of pro-apoptotic genes such as Bid (BH3 interacting domain death agonist protein), Bad (Bcl-2 associated agonist of cell death protein) and Bim that activate the intrinsic apoptotic pathway [42,58,63,64].
It can be concluded that in tumor cells exposed to HDACs inhibitors pro-apoptotic genes involved in the extrinsic (TRAIL, DR5, FAS, FAS-L, and TNF-α) and/or intrinsic apoptotic pathways (BAX, BAK and APAF1) are up-regulated, while anti-apoptotic genes (Bcl-2 and XIAP (X-linked inhibitor of apoptosis protein)) are downregulated [10]. HDAC inhibitors can, however, enhance the levels of anti-apoptotic protein Bcl-2 via activation of ERK [65]. Besides, these effects on gene expression, the HDAC inhibitors increase amounts of reactive oxygen species (ROS) that can induce apoptosis in leukemic cells (Jurkat, ML-1, U937, HL-60, K-562, CEM-CCRF and its doxorubicin selected P-glycoprotein overexpressing subline and FDC-P1 and sublines overexpressing Bid, Bcl-2 and Bid plus Bcl-2) [63,66,67]. SAHA and entinostat increase expression of binding protein-2 (TBP-2) that inhibits thioredoxin in LNCaP prostate cancer, T24 bladder cancer and MCF7 breast cancer cells [68]. Thioredoxin is an intracellular antioxidant, therefore treatment of tumor cells by these HDAC inhibitors induce ROS-dependent apoptosis [69,70]. We found that VPA induces apoptosis more effectively under hypoxic conditions and overcomes hypoxia-induced resistance to cisplatin (CDDP) in high risk neuroblastoma derived cells UKF-NB-3 and CDDP resistant subline [71] probably by induction of HIF-1α degradation [72].
4.3. The Effects on Induction of Autophagy
Acetylation of many autophagy-related proteins, such as the product of the autophagy-related genes (ATGs), is regulated by the balance between HATs and HDACs [73]. Autophagy can also be regulated through the acetylation of transcription factors such as FOXO [74]. Different HDACs influence autophagic activity by different mechanisms as showed the results of several experiments described below. HDAC6 induces autophagy when the ubiquitin-proteasome system (UPS) is impaired. Knockdown of HDAC2 inhibits autophagy in cardiomyocytes [75]. On the contrary, inhibition or knockdown of HDAC1 in HeLa cells promote formation of autophagic vacuoles [76]. HDAC10 induces autophagy-mediated cell survival in neuroblastoma (E(2)-C, Kelly, and IMR32 cells) and its inhibition sensitizes cells to cytostatics [77]. SIRT1 forms a complex with components of the autophagy machinery (Atg5, -7, and -8) and stimulates autophagy [78].
The role of HDAC inhibitors induced autophagy in the death of cancer cells is still controversial. Some studies have reported that autophagy serves as a cell death mechanism as either autophagy inhibitors or knockdown of ATGs reduces the anticancer effect of HDAC inhibitors. Moreover, using different in vivo models, a combination of HDAC inhibitors and autophagy inhibition reduces colon cancer cells HCT116 growth [79]. In contrast, autophagic degradation of intracellular components is a death signal and leads to the cytotoxic effect of autophagy. For example, in panel of hepatocellular carcinoma cells SAHA-induced cytotoxicity is inhibited by 3-methyladenine, which inhibits autophagy by blocking the autophagosome formation via the inhibition of class III PI3K, or by Atg5 knockout [80]. Cell death in endometrial stromal sarcoma cells induced by SAHA is caused by autophagy [81]. SAHA induces apoptosis in TP53 wild type cancer cells, while the absence or degradation of cytoplasmatic p53 leads to activation of the autophagic pathway which consequently induces cell death [82]. The above-mentioned discrepancies might be due to differences in the used models, cancer cells, HDAC inhibitors and their doses.
Several signaling pathways play a role in the induction of autophagy by HDAC inhibitors. mTOR (Mechanistic target of rapamycin) is one of the most important suppressors of autophagy via phosphorylation and inactivation of the ULK1 (Unc 51 like autophagy activating kinase 1) complex which is an upstream component of the autophagy pathway. mTOR inactivation by SAHA restores functions of ULK1 [80,83,84]. Overexpression of ATG genes induced by SAHA is caused by the stimulation of NF-κB activity via modulation of RelA/p65 (NF-κB p 65 subunit) signaling [85]. Some studies also show that SAHA cause autophagy by ROS production in leukemic and hepatocellular carcinoma derived cells [84,86]. The fact that some HDAC inhibitors can induce cell death in the apoptosis-resistant cells by induction of autophagy seems to be the important feature for clinical practice. Romidepsin and HDAC1 siRNA induce autophagy in HeLa cells [76]. SAHA inhibits growth of short term culture glioblastoma cells xenografts in nude mice by autophagy induction via downregulation of AKT-mTOR signaling [87]. Based on the above-mentioned facts, we can suppose that induction of autophagy by HDAC inhibitors may be a promising therapeutic anticancer strategy.
4.4. The Effects on Non-Coding RNA
HDAC inhibitors have been described to alter non-coding RNA expression. TSA and SAHA induce miR-129-5p overexpression and apoptosis in thyroid cancer cells. miR-129-5p alone induces cell death and knockdown experiments showed that its expression is necessary for HDAC inhibitor-induced cell death [88]. Treatment of breast cancer cells and normal human fibroblasts with a combination of sodium butyrate and panobinostat increased the expression of miR-31 which induces cellular senescence via inhibition of BIM1 (Bisindolylmaleimide-based protein kinase C inhibitor 1) [89]. This was demonstrated by transfection of cells by vector overexpressing BMI1 and on the other hand by its knock down by BMI1 shRNA. HDAC inhibition in B-cell lymphomas cells (Daudi, Ramos, Raji, Su-DHL-6, OCI-Ly-19 and OCI-Ly-3) and non-malignant B cells silences Myc-mediated transcriptional repression of the miR-15 and let-7 miRNA families, which induces apoptosis by downregulation of the anti-apoptotic genes Bcl-2 and Bcl-xL and, thus, activates apoptotic pathways [90]. HDAC1 enhances miRNA processing by acetylating protein DGCR8 (DiGeorge critical region 8 protein) which processes miRNA and this acetylation increases the production of mature miRNAs in embryonic kidney cells and acute myeloid leukemia cells (AML) [91]. Moreover, several HDACs are also targeted by miRNA, e.g., miR-449a regulates prostate cancer both androgen dependent and independent cells (PC-3, DU-145, BHP-1 and LNCaP) growth and viability by targeting and repressing HDAC1 [92].
Long non-coding RNAs (lncRNAs) are important cellular regulator molecules at the transcriptional and post-transcriptional level. They exhibit a wide spectrum of functions such as regulation of alternative splicing, transcriptional patterns, and protein activity. They also have epigenetic effects and are precursors of miRNAs [93]. One of the mechanisms of transcription regulation by lncRNAs is recruiting histone modifying complexes (e.g., PRC (Polycomb repressive complex)) to target loci which are either activated or silenced, depending on the histone marks [94].
The data of Yang et al. showed that approximately 5% of intergenic lncRNA and about 6% of protein coding genes were significantly differentially expressed in hepatocellular cancer cell lines (Huh7, Bel7402, Bel7721, and HepG2) after treatment with TSA [95]. Intergenic lncRNAs regulate gene expression through modifying the chromatin complexes or RNA binding proteins and their aberrant expression is associated with cancer [96].
Low expression of lncRNA Xist (X inactive specific transcript which plays a major role in the X inactivation process) seems to be a predictor of efficacy of abexinostat in breast cancer cells [97]. One may speculate from the above-mentioned facts that changes of lncRNAs expression may be one of the mechanisms of the anticancer effect of HDAC inhibitors. However, further studies with non-coding RNAs are necessary.
4.5. The Effects on Cellular Signaling Pathways
Another mechanism of anticancer effects of HDAC inhibitors is regulation of cell differentiation by activation of some of protein kinases (i.e., ERK). Protein kinases modulate biological processes like cell growth, differentiation and apoptosis. HDAC inhibitors were found to increase DNA binding and transactivation activity of AP-1 transcription factor via ERK activation, increase expression of c-Jun (Jun activation domain binding protein) and induce its phosphorylation in SH-SY5Y neuroblastoma cells [65,98]. Although it is not yet clear how HDAC inhibitors affect ERK, it is supposed that they can induce synthesis of a still unknown factor that activate the ERK signaling pathway [65] or is incorporated into phospholipid molecules that activates ERK via the phosphatidylinositol 3-kinase (PI-3K)/Janus kinase 2 (JAK 2)/MEK-1-dependent and the tyrosine kinase-Ras-dependent pathways [99,100]. HDAC inhibitors also increase the expression of genes that are involved in regulation of ERK/AP-1 signaling, for example the growth associated protein-43 (GAP-43) and Bcl-2, and hence they might increase growth of some cancer cells, e.g., in SH-SY5Y neuroblastoma cells or immature cortical neuroblasts in GAP-43−/− mice [65,101].
VPA also affects Wnt signaling that is due to phosphorylation of serine 9 in the glycogen synthase kinase-3β (GSK-3β) [102]. The Wnt signaling pathway plays an important role in various cancers such as colon, breast, ovarian, prostate and endometrial cancers as well as medulloblastoma and melanoma [103]. Inactivation of APC (Adenomatous polyposis coli) functions and of β-catenin induces overexpression of the HDAC2 which protects colorectal cancer cells HT-29 from death [10]. Therefore, we may speculate that HDAC2 is a target of APC/β-catenin. Moreover, HDAC inhibitors decrease polyp generation in a colon carcinoma model of APC deficient mice, probably via damage of the HDAC2 by degradation in proteasomes [10]. Furthermore, VPA increases proliferation and self-renewal of normal hematopoietic stem cells by inhibition of GSK-3β that activates the Wnt pathway [104].
Acetylation of p53, which may be caused by HDAC inhibitors, decreases generation of a complex p53/Mdm2 E3 ligase, whereas hypoacetylation increases its degradation by the proteasome and cancels p53-mediated growth arrest and apoptosis as demonstrated by experiments with human non-small cell carcinoma cells H1299 transfected by different Tp53 mutants [48]. In addition, acetylation induced by HDAC inhibitors influences the expression of several proteasomal enzymes (Ubc8 E2 ubiquitin conjugase, RLIM-subunit of the SCF E3 family of the ubiquitin ligase) in human embryonal kidney cells [105].
4.6. The Antiangiogenic Effect
HDAC inhibitors can affect cancer angiogenesis and inhibit cellular stress response pathways, thereby interfering with the metastatic process. The anti-angiogenic effects are associated with down-regulation of pro-angiogenic genes such as the genes of the vascular endothelial growth factor (VEGF) and/or endothelial nitric oxide synthase (eNOS) [106,107,108]. Expression and enzymatic activity of eNOS are influenced by its phosphorylation which is catalyzed by Akt and numerous hemodynamic and hormonal stimuli [109,110,111]. HDAC inhibitors decrease the stability of eNOS mRNA by binding to the 5′-untranslated region of eNOS mRNA [112]. In addition, HDAC inhibitors decrease the levels of the VEGF receptors in neuroblastoma cells [113].
Moreover, HDAC inhibitors induce hyperacetylation of HIF-1α, a pro-angiogenic transcription factor, which induce its degradation [72]. VPA decreases angiogenesis by enhancing production of the anti-angiogenic proteins thrombospondin-1 and activin A via downregulation of pro-angiogenic factors such as the basic fibroblast growth factor (bFGF) [114]. Expression array showed that in prostate cancer cells VPA induces the up-regulation of TSP1 (Thrombospondin-1), multiple TIMP (Tissue inhibitor of metalloproteinase) isoforms and TGFβ (Transforming growth factor β), while expression of IGF1 (Insulin like growth factor 1) and VEGF is decreased [115]. VPA and TSA decrease the formation of capillary tubes of HUVEC (human vascular endothelial cells), but they do not induce their apoptosis [116].
On the contrary, Lin et al. found that HDAC inhibition resulted in the development of metastatic phenotype including increasing of matrix metalloproteinases (MMPs) expression in some cancer cell lines (liver (SNU-398, Huh6, HepG2, Hep3B, Huh7, PLC5, HCC36, TONG, HA59T, Sk-hep-1, HA22T, and Malhavu), lung (H358, H1437, H661, H226Br, H1299, CL1-3, CL1-0, H23, H928, and A549), gastric (NUGC, SC-M1, AZ521, AGS, and HR), and breast (MDA-231, Hs578T, and MCF-7) cancer) [117]. Degradation of extracellular matrix by MMPs is important for angiogenesis, tissue invasion and metastasis.
4.7. HDAC Inhibitor-Induced Modulation of Immune Response
A decrease in HDAC activity alters expression of MHC (Major histocompatibility complex) and costimulatory molecules [118,119]. The subsequent increase in immunogenicity intensifies activation of T cells and the prolonged survival of experimental animals [120,121]. Moreover, inhibition of HDAC6 activates naïve T cells [119,120]. In addition, HDAC inhibitors influence different lymphocyte populations. Inhibition of Class II HDACs enhances number and function of Treg and Class I HDAC inhibitors enhances the functions of NK cells and CD8 T cells [122].
After exposure to SAHA or entinostat breast (MDA-MB-231), prostate (LNCaP) and pancreas (AsPC-1) carcinoma cells are more sensitive to T-cell-mediated lysis in vitro [123]. This increased immune reaction is directed against the HLA (Human leukocyte antigens) class I/epitope complexes and the increased sensitivity to antigen-specific cytotoxic T-lymphocyte lysis indicate that HDAC inhibition induces immunogenic modulation by promoting a signature of immune recognition. Several tumor associated antigens have been shown to be epigenetically silenced in malignancies impede immune recognition by cytotoxic T cells and contributing to worse prognosis [124,125]. The exposure of human carcinoma cells to SAHA has previously been shown to result in upregulation of HLA related genes [126].
4.8. The Effects on Stem Cells
Epigenetic changes are important for reprogramming of somatic cells into pluripotent stem cells. Therefore, several inhibitors of epigenetic-modifying enzymes including HDAC are able to reprogram somatic cells into the pluripotent stem cells by modifying a chromatin structure and making it more permissive to transcription factors [127]. There was described amplification and maintenance of normal human hematopoietic stem cells induced by HDAC inhibitors [128], enhancement of the epithelial–mesenchymal transition of colorectal and breast cancer cells [129,130] and induction of CD133 (a marker of cancer stem cells in some cancers including the brain tumors) expression in human glioma [131]. The experimental in vitro study showed that HDAC3 promoted self-renewal of glioma stem cells and those experimental results were proved also by the studies utilizing the tumor samples [132]. Our recent study shows that HDAC inhibitors (VPA, SAHA, and MS-275) increase CD133 significantly in neuroblastoma cell lines that do not show methylated its CpG promoters and VPA treatment may increase CD133+ cells that show higher resistance to cytostatics than CD133− cells. This increase, found in the CD133+ cells was not caused by elimination of CD133− cells. Moreover, VPA treatment enhanced clonogenicity and the ability to generate neurospheres, increased Akt phosphorylation and induced expression of the pluripotency transcription factors (Oct-4 (octamer-binding transcription factor 4), Nanog (Homeobox Transcription Factor Nanog), Sox2 (sex determining region Y)) [133]. Similar increase of CD 133+ cells was found also in glioblastoma cells treated by VPA [134]. Amplification of cancer “stem like cells” might be unwanted action of HDAC inhibitors.
4.9. Other Effects of HDAC Inhibitors
Several HDAC inhibitors are known to be metabolized by cytochrome P450 (CYP) and they also influence expression of the CYP enzyme proteins and their enzyme activities. Since CYP enzymes are involved in biosynthesis and metabolism of many endogenous physiologically active substances and drugs, this mechanism may be involved in potentiating of the effects of some anticancer drugs by HDAC inhibitors. We found that VPA and TSA change CYP enzyme expression in neuroblastoma cells [135].
Several proteins important for DNA repair e.g., Ku70, flap structure-specific endonuclease 1 (FEN1), Werner syndrome (WRN), ataxia teleangiectasia mutated protein (ATM), mediator of DNA damage checkpoint 1 (MDC1) and DNA-dependent protein kinases (DNA PK) are regulated by acetylation which may be increased by HDAC inhibitors [136]. HDAC1 and 2 inhibition decreases the DNA repair processes mediated by BAL-associated protein (BBAP) which protects cells against DNA-damaging agents [137]. HDAC6 and SIRT2 are able to deacetylate α-tubulin and so stabilize microtubules [138].
HDAC inhibitors have also anti-parasitic activity, e.g., against Plasmodium and Trypanosoma [139].
5. Combination of HDAC Inhibitors with Other Anticancer Therapeutic Modalities—Preclinical Studies
The results found in the in vitro and in vivo experiments using various cancer cells have demonstrated that combination of HDAC inhibitors with anticancer drugs and/or radiotherapy have synergistic or additive effects [37,140] (see Table 3). Therefore, combinations chemotherapy and/or radiotherapy with HDAC inhibitors have also been used in clinical trials [141].
HDAC inhibitors were combined with other epigenetic modifiers. Inhibitors of DNA methyl transferases azacitidine and decitabine had increased antitumor effects on myelodysplastic syndrome, prostate, ovarian and pancreatic endocrine tumors cells when used with HDAC inhibitors [52,55,142,159,160]. Decitabine and VPA both induced apoptosis and the combination increased their effects both in vitro and in vivo on leukemic cells and on medulloblastoma and rhabdomyosarcoma that develop in Ptch1 (Protein patched homolog 1) knockout mice [143,144]. On the contrary, this combination induced the CD133 expression in neuroblastoma cells with the methylated CD133 promotor [133]. Co-treatment of several cancer cells (prostate, pancreatic, lung and AML) with TSA and decitabine synergistically induced apoptosis [52,55,145,146]. In addition, tranylcypromine (monoamine oxidase inhibitor) and SAHA showed synergistic enhancement of apoptosis in glioblastoma cells [58].
Positive effects have been reported for combinations of HDAC inhibitors and ROS-generating agents. Adaphostin (a tyrosine kinase inhibitor that induces intracellular ROS) potentiates entinostat and SAHA induced apoptosis in leukemia cells and depletion of ROS scavenger GSH potentiates the anti-leukemic effect of SAHA [58,147]. Panobiostat sensitized lung adenocarcinoma cells including cells with K-ras (Kirsten rat sarcoma viral oncogene homolog) or epidermal growth factor receptor (EGFR) mutation to the anti-proliferative effects of the tyrosine kinase inhibitor erlotinib in the in vitro experiment [148]. EZH2 (catalytic subunit of polycomb repressive complex 2) interacts with class I HDACs and transcriptional repression by EZH2 requires the activity of the HDACs and HDAC downregulates PRC2 proteins. Therefore, one may speculate that concurrent inhibition of these epigenetic silencing enzymes has synergistic antitumor effects. Co-treatment non-small cell lung cancer (NSCLC) cells with 3-deazaneplanocin A (EZH inhibitor) and vorinostat synergistically induced apoptosis in all tested NSCLC cell lines, independently on their EGFR status. The co-treatment by EZH and HDAC inhibitors induced accumulation of p27Kip (Cyclin dependent kinase inhibitor p27) and a decrease in cyclin A, suppressed EGFR signaling, both in EGFR-wild-type and mutant NSCLC cells [149].
Another effective combination of HDAC inhibitors is that with proteasome inhibitors. Cancer cell death due to a combination of proteasome and HDAC inhibitors is caused by induction of oxidative stress, endoplasmic reticulum stress and stimulations of JNK (Jun-N-terminal kinase). Treatment of multiple myeloma cells with bortezomib made the cells more sensitive to HDAC inhibitors [150]. Trials in patients with multiple myeloma demonstrated an increase in SAHA antitumor effects in combination with bortezomib [161,162]. Proteasome inhibitor marizomib potentiates apoptosis induced by SAHA or entinostat in pancreatic cancer cells [151]. The in vitro and in vivo studies with lymphomas cells including bortezomib resistant ones showed that proteasome inhibitor carfilzomib increased the anticancer effect of SAHA [152,153].
Numerous studies show synergisms or additive effects of HDAC inhibitors and DNA damaging agents such as topoisomerase inhibitors, DNA intercalators, inhibitors of DNA synthesis, covalently modifying DNA agents (i.e., doxorubicin, epirubicin, etoposid, CDDP, melphalan, and temozolomide) and ionizing radiation in many cancer cell lines (reviewed in [37]). VPA combined with CDDP or etoposide (VP-16), but not with vincristine, was found to act synergistically. These results indicate that HDAC inhibitors increase the cytotoxic efficiency of the only of several DNA-damaging anticancer drugs. The mechanisms of the potentiating effects of HDAC inhibitors have not yet been fully explained. The sequence of drug application is important for sensitizing neuroblastoma cells to CDDP and VP-16 by VPA. It potentiates the cytotoxic effect of CDDP or VP-16 only when added simultaneously, or when cells were preincubated with cytostatics before exposure to HDAC inhibitors. In contrast, the reversed sequence (pretreatment of cells with VPA) did not give any further increase in cytotoxicity of cytostatics [154]. The results found by Luchenko et al. [155] indicated that DNA relaxation is not required for the synergy of belinostat and romidepsin with CDDP and VP-16 in small cell lung cancer cells. One can speculate that the changes in the structure of DNA caused by CDDP and VP-16 (formation of DNA adducts or DNA cross-links by CDDP, intercalation of VP-16 into DNA, and formation of ROS by both drugs) increase accessibility of nucleosomal core histones to their acetylation, which additionally determines transcription of some genes involved in DNA repair or apoptosis. Kim et al. described that pre-treatment of cells (glioblastoma lines D54 and U118, breast cancer MCF-7, normal breast MCF-12F and intestinal FHs74Int) with SAHA or TSA enhances the cytotoxicity of VP-16, ellipticine, doxorubicin, and CDDP but not that of drugs which do not target the DNA, such as 5-fluorouracil [156]. It only partially agreed with our results, Kim et al. [156] found potentiation of cytotoxicity after pretreatment of cells with HDAC inhibitors before exposure to other drugs, while the results of our experiments indicated the opposite phenomenon; the administration of VPA after DNA-damaging cytostatic increased their cytotoxicity [154]. Nevertheless, both groups used different HDAC inhibitors and different cell lines.
HDAC inhibitors are able to deacetylate α-tubulin which has stabilizing effect on microtubules. There was found that combination of TSA and paclitaxel increases apoptosis induction in endometrial and anaplastic thyroid carcinoma cells [138,157].
The in vitro experiments and the experiments with mice in vivo showed that the combination of VPA and temozolomide enhanced the apoptotic and autophagic cell death, as well as suppressed the migratory activities in temozolomide-resistant glioblastoma cells that express O-(6)-methylguanine-DNA methyltransferase (MGMT). The effect of VPA is caused by downregulation of MGMT [158].
6. Clinical Studies and Registered Drugs
As mentioned above, several HDAC inhibitors are tested in clinical studies and four of them are approved drugs (vorinostat, belinostat, panobinostat and romidepsin). The most common toxicities of those HDAC inhibitors, which are not the class-specific and have been observed in all their types, are thrombocytopenia, neutropenia, diarrhea, nausea, vomiting and fatigue [163].
SAHA had only the modest activity as a single agent (response rate is 10–20% in AML and myelodysplastic syndrome (MDS)). However, a combination with 5-azacitidine increased its response rate by 30%. The combination of SAHA with idarubicin and cytarabine had synergistic activity that was maximal when SAHA preceded cytarabine and there were responses in all patients with AML with FLT3/ITD mutation, which is otherwise associated with a worse prognosis [164,165]. Administration of SAHA in refractory CTCL patients showed in phase II trials objective response in nearly 30% [166,167]. HDAC inhibitor silences the genes in common translocations associated with hematological malignancies, e.g., those of the AML/ETO fusion protein [168]. Phase I study of patients with advanced leukemia and MDS treated with SAHA showed clinical benefit in AML and MDS [169]. The clinical phase II study proved that panobinostat is effective in relapsed/refractory Waldenstrom macroglobulinemia [170]. Mocetinostat had the only limited efficacy as showed the clinical study phase II for the treatment of patients with refractory chronic lymphocytic leukemia (CLL). Therefore, the combination with other agents such as conventional chemotherapeutic drugs was recommended [171]. Panobinostat underwent phase I and II clinical studies for the treatment of both solid and hematologic malignancies and phase III clinical trials for CTCL and chronic myeloid leukemia that showed promising results against CTCL [172] and leukemias, respectively, and proved the increased acetylation of histones in malignant cells that was associated with apoptosis [173]. Panobinostat also underwent phase III clinical trials against CTCL and leukemia in an oral form and showed the positive effect. Despite the promising results in the treatment of CTCL, SAHA and romidepsin have not been effective in studies with different solid tumors (neuroendocrine tumors, glioblastoma multiforme, mesothelioma, refractory breast, colorectal, NSCLC, prostate, head and neck, renal cell, ovarian, cervical and thyroid cancers) and Hodgkin lymphoma [174,175].
Therefore, the clinical trials tend to combine HDAC inhibitors with other drugs to enhance their anticancer effects. A clinical study showed that VPA increased efficacy of radiochemotherapy with temozolomide in glioblastoma patients [176]. VPA with doxorubicin appeared to have the 16% response rate in patients suffering from refractory or recurrent mesothelioma [177]. SAHA improved effect of carboplatin and paclitaxel in NSCLC [178] and reversed resistance to tamoxifen in estrogen receptor positive breast cancer [179]. Phase II study with a combination of erlotinib with entinostat showed prolonged progression-free survival in NSCLC patients harboring high E-cadherin levels, irrespective of the EGFR genotype compared to erlotinib monotherapy, while does not improve the outcome of patients with low E-cadherin [180]. Clinical trial with the combination of the immune checkpoint therapy with the HDAC inhibitor is ongoing [181].
7. Conclusions and Future Direction
HDACs are involved in different cellular pathways and functions; nevertheless, further studies are necessary to disclose all their functions and cellular interactions, which might result in development of more efficient therapy with HDAC inhibitors. HDAC inhibitors seem to be a promising group of anti-cancer drugs, particularly in combination with other anti-cancer drugs and/or radiotherapy. Of HDAC inhibitors, SAHA and romidepsin have been approved for CTCL, romidepsin also for PTCL, belinostat for therapy of PTCL and panobinostat for multiple myeloma. Many other HDAC inhibitors are in clinical trials for the treatment of both hematological and solid malignancies. Even though many biological effects of HDAC inhibitors have been found, explanations of these effects remain unclear.
In addition, their use in the combination with other drugs and the schedule of such drug combinations need to be investigated, in both preclinical and clinical studies. Indeed, recently, we have found that sequence of HDAC inhibitor and DNA-damaging drug is important for increase of cytotoxicity [154]. The discovery of predictive factors for evaluation of HDAC inhibitors is also necessary as demonstrated in the study with combination of erlotinib with entinostat [180]. The other most important question is whether the pan-HDAC inhibitors or the selective inhibitors will be more efficient in different types of cancers. Furthermore, assumption about the role of some HDAC inhibitors as inducers of cancer stem cells [133,134] or about the phenomenon that HDAC inhibition may enhance the epithelial–mesenchymal transition of cancer cells [129,130] needs further studies. We may conclude that while the preliminary results are promising, large multicentric clinical studies are needed to ascertain whether this treatment offers beneficial clinical outcomes with tolerable side-effect profiles.
However, the great potential for epigenetic therapies that is caused by the fact that epigenetic changes are reversible might be considered.
Acknowledgments
This work was supported by grant GA CR 17-12816S and by the Ministry of Health of the Czech Republic for conceptual development of research organization 00064203 (University Hospital Motol, Prague, Czech Republic).
Author Contributions
Tomas Eckschlager and Jan Hrabeta wrote the manuscript; Johana Plch contributed to the figure and table design and the literature research; and Marie Stiborova revised and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, G.; Margueron, R.; Hu, G.; Stokes, D.; Wang, Y.H.; Reinberg, D. Highly compacted chromatin formed in vitro reflects the dynamics of transcription activation in vivo. Mol. Cell 2010, 38, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Scarpati, G.D.V.; Di Lorenzo, G.; Pisconti, S. Epigenetic control of gene expression: Potential implications for cancer treatment. Crit. Rev. Oncol. Hematol. 2017, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Herman, J.G.; Guo, M. Epigenome-based personalized medicine in human cancer. Epigenomics 2015, 8, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and nuclear recruitment of HDACl in hormone refractory prostate cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.M.J.; Johnstone, R.R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.-I.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; Von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Horiuchi, A.; Kikuchi, N.; Hayashi, T.; Fuseya, C.; Suzuki, A.; Konishi, I.; Shiozawa, T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int. J. Cancer 2010, 127, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Allis, C.D. Histone acetylation and chromatin assembly: A single escort, multiple dances? Cell 1996, 87, 5–8. [Google Scholar] [CrossRef]
- Kazanets, A.; Shorstova, T.; Hilmi, K.; Marques, M.; Witcher, M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as epigenetic regulators of the immune system: Impacts on cancer therapy and inflammatory diseases. BioMed Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yan, H.; Zhuang, S. Histone deacetylases as targets for treatment of multiple diseases. Clin. Sci. 2013, 124, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [PubMed]
- Trapp, J.; Jung, M. The role of NAD+ dependent histone deacetylases (sirtuins) in ageing. Curr. Drug Targets 2006, 7, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, P.; Wong, J. HDAC3: Taking the SMRT-N-CoRrect road to repression. Oncogene 2007, 26, 5439–5449. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Wang, J.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Mariadason, J.M.; Corner, G.A.; Augenlicht, L.H. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: Comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. 2000, 60, 4561–4572. [Google Scholar] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Lavu, S.; Boss, O.; Elliott, P.J.; Lambert, P.D. Sirtuins—Novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 2008, 7, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Valente, S.; Mai, A. Small-molecule inhibitors of histone deacetylase for the treatment of cancer and non-cancer diseases: A patent review (2011–2013). Expert Opin. Ther. Pat. 2014, 24, 401–415. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, W.K.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors in cancer therapy. Expert Opin Investig. Drugs 2007, 16, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Eckschlager, T.; Poljakova, J.; Hrabeta, J.; Adam, V.; Kizek, R.; Frei, E. The synergistic effects of DNA-targeted chemotherapeutics and histone deacetylase inhibitors as therapeutic strategies for cancer treatment. Curr. Med. Chem. 2012, 19, 4218–4238. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Kretsovali, A.; Hadjimichael, C.; Charmpilas, N. Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int. 2012, 2012, 184154. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Sung, J.; Cohen, M.; Chowdhury, W.H.; Sachs, M.D.; Li, Y.; Lakshmanan, Y.; Yung, B.Y.; Lupold, S.E.; Rodriguez, R. Valproic acid inhibits invasiveness in bladder cancer but not in prostate cancer cells. J. Pharmacol. Exp. Ther. 2006, 319, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Stockhausen, M.-T.; Sjölund, J.; Manetopoulos, C.; Axelson, H. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br. J. Cancer 2005, 92, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Vrana, J.A.; Decker, R.H.; Johnson, C.R.; Wang, Z.; Jarvis, W.D.; Richon, V.M.; Ehinger, M.; Fisher, P.B.; Grant, S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 1999, 18, 7016–7025. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent G1arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: Signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Gius, D.; Cui, H.; Bradbury, C.M.; Cook, J.; Smart, D.D.K.; Zhao, S.; Young, L.; Brandenburg, S.A.; Hu, Y.; Bisht, K.S.; et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 2004, 6, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lu, S.; Wu, L.; Chai, G.; Wang, H.; Chen, Y.; Sun, J.; Yu, Y.; Zhou, W.; Zheng, Q.; et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol. Cell. Biol. 2006, 26, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Mahyar-Roemer, M.; Roemer, K. p21 Waf1/Cip1 can protect human colon carcinoma cells against p53-dependent and p53-independent apoptosis induced by natural chemopreventive and therapeutic agents. Oncogene 2001, 20, 3387–3398. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yokozaki, H.; Kuniyasu, H.; Hayashi, K.; Naka, K.; Ono, S.; Ishikawa, T.; Tahara, E.; Yasui, W. Effect of trichostatin A on cell growth and expression of cell cycle- and apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int. J. Cancer 2000, 88, 992–997. [Google Scholar] [CrossRef]
- Qiu, L.; Burgess, A.; Fairlie, D.P.; Leonard, H.; Parsons, P.G.; Gabrielli, B.G. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, D.; Donadelli, M.; Pozza, E.D.; Rinalducci, S.; Zolla, L.; Scupoli, M.T.; Righetti, P.G.; Scarpa, A.; Palmieri, M. Synergistic effect of trichostatin A and 5-aza-2′-deoxycytidine on growth inhibition of pancreatic endocrine tumour cell lines: A proteomic study. Proteomics 2009, 9, 1952–1966. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Li, Q.L.; Ito, K.; Sakakura, C.; Fukamachi, H.; Inoue, K.I.; Chi, X.Z.; Lee, K.Y.; Nomura, S.; Lee, C.W.; Han, S.B.; et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002, 109, 113–124. [Google Scholar] [CrossRef]
- Walton, T.J.; Li, G.; Seth, R.; McArdle, S.E.; Bishop, M.C.; Rees, R.C. DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor β and induce apoptosis in prostate cancer cell-lines. Prostate 2008, 68, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Singh, M.M.; Rivera-Del Valle, N.; Manton, C.A.; Chandra, J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J. Biomed. Biotechnol. 2011, 2011, 514261. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr. Cancer Drug Targets 2008, 8, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Ahn, S.H.; Kim, Y.K.; Bae, G.U.; Yoon, J.W.; Hong, S.; Lee, H.Y.; Lee, Y.W.; Lee, H.W.; Han, J.W. Apicidin, a Histone Deacetylase Inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 2002, 277, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA. 2001, 98, 10833–10838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tan, J.; Zhuang, L.; Jiang, X.; Liu, E.T.; Yu, Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 2005, 102, 16090–16095. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.X.; Huang, L.D.; Jiang, Y.M.; Gutkind, J.S.; Manji, H.K.; Chen, G. The Mood Stabilizer Valproic Acid Activates Mitogen-activated Protein Kinases and Promotes Neurite Growth. J. Biol. Chem. 2001, 276, 31674–31683. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Mobley, A.; Miller, C.; Boklan, J.; Chandra, J. Potentiation of reactive oxygen species is a marker for synergistic cytotoxicity of MS-275 and 5-azacytidine in leukemic cells. Leuk. Res. 2008, 32, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Butler, L.M.; Zhou, X.; Xu, W.-S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar] [PubMed]
- Shao, L.E.; Diccianni, M.B.; Tanaka, T.; Gribi, R.; Yu, A.L.; Pullen, J.D.; Camitta, B.M.; Yu, J. Thioredoxin expression in primary T-cell acute lymphoblastic leukemia and its therapeutic implication. Cancer Res. 2001, 61, 7333–7338. [Google Scholar] [PubMed]
- Cipro, Š.; Hřebačková, J.; Hraběta, J.; Poljaková, J.; Eckschlager, T. Valproic acid overcomes hypoxia-induced resistance to apoptosis. Oncol. Rep. 2012, 27, 1219–1226. [Google Scholar] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 3885–3901. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Choi, I.K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Oehme, I.; Linke, J.-P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Hrzenjak, A.; Kremser, M.L.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: Evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 68. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Autophagy, B.; Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, M.; Van Grevenynghe, J.; Belgnaoui, S.M.; Nguyên, L.; Di Lenardo, T.; Semmes, O.J.; et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar]
- Park, M.A.; Reinehr, R.; Haussinger, D.; Voelkel-Johnson, C.; Ogretmen, B.; Yacoub, A.; Grant, S.; Dent, P. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol. Cancer Ther. 2010, 9, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Chiao, M.T.; Cheng, W.Y.; Yang, Y.C.; Shen, C.C.; Ko, J.L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Brest, P.; Lassalle, S.; Hofman, V.; Bordone, O.; Tanga, V.G.; Bonnetaud, C.; Moreilhon, C.; Rios, G.; Santini, J.; Barbry, P.; et al. MiR-129–5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells. Endocr. Relat. Cancer 2011, 18, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Dimri, M.; Dimri, G.P. MicroRNA-31 is a transcriptional target of histone deacetylase inhibitors and a regulator of cellular senescence. J. Biol. Chem. 2015, 290, 10555–10567. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Hiebert, S.W.; Eischen, C.M. Myc Induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 2016, 76, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Kikuchi, J.; Furukawa, Y. Histone deacetylase 1 enhances microRNA processing via deacetylation of DGCR8. EMBO Rep. 2012, 13, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Moran, V.A.; Perera, R.J.; Khalil, A.M. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012, 40, 6391–6400. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N. Noncoding RNA and Polycomb recruitment. RNA 2013, 19, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhong, Y.; Xie, H.; Lai, X.; Xu, M.; Nie, Y.; Liu, S.; Wan, Y.J.Y. Induction of the liver cancer-down-regulated long noncoding RNA uc002mbe.2 mediates trichostatin-induced apoptosis of liver cancer cells. Biochem. Pharmacol. 2013, 85, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Salvador, M.A.; Wicinski, J.; Cabaud, O.; Toiron, Y.; Finetti, P.; Josselin, E.; Lelièvre, H.; Kraus-Berthier, L.; Depil, S.; Bertucci, F.; et al. The histone deacetylase inhibitor abexinostat induces Cancer stem cells differentiation in breast Cancer with low Xist expression. Clin. Cancer Res. 2013, 19, 6520–6531. [Google Scholar] [CrossRef] [PubMed]
- Asghari, V.; Wang, J.F.; Reiach, J.S.; Young, L.T. Differential effects of mood stabilizers on FosrJun proteins and AP-1 DNA binding activity in human neuroblastoma SH-SY5Y cells. Mol. Brain Res. 1998, 58, 95–102. [Google Scholar] [CrossRef]
- Bassa, B.V.; Roh, D.D.; Vaziri, N.D.; Kirschenbaum, M.A.; Kamanna, V.S. Lysophosphatidylcholine activates mesangial cell PKC and MAP kinase by PLCgamma-1 and tyrosine kinase-Ras pathways. Am. J. Physiol. 1999, 277, F328–F337. [Google Scholar] [PubMed]
- Cieslik, K.; Abrams, C.S.; Wu, K.K. Up-regulation of endothelial nitric-oxide synthase promoter by the phosphatidylinositol 3-kinase γ/Janus kinase 2/MEK-1-dependent pathway. J. Biol. Chem. 2001, 276, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Shen, Y.; Schaefer, J.; Meiri, K.F. Failure to express GAP-43 during neurogenesis affects cell cycle regulation and differentiation of neural precursors and stimulates apoptosis of neurons. Mol. Cell. Neurosci. 2001, 17, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Blaheta, R.A.; Cinatl, J. Anti-tumor mechanisms of valproate: A novel role for an old drug. Med. Res. Rev. 2002, 22, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Bug, G.; Gul, H.; Schwarz, K.; Pfeifer, H.; Kampfmann, M.; Zheng, X.; Beissert, T.; Boehrer, S.; Hoelzer, D.; Ottmann, O.G.; Ruthardt, M. Valproic acid stimulates proliferation and self-renewal of hematopoietic stem cells. Cancer Res 2005, 65, 2537–2541. [Google Scholar] [CrossRef] [PubMed]
- Krämer, O.H.; Zhu, P.; Ostendorff, H.P.; Golebiewski, M.; Tiefenbach, J.; Peters, M.A.; Brill, B.; Groner, B.; Bach, I.; Heinzel, T.; et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003, 22, 3411–3420. [Google Scholar] [CrossRef] [PubMed]
- Zupkovitz, G.; Tischler, J.; Posch, M.; Sadzak, I.; Ramsauer, K.; Egger, G.; Grausenburger, R.; Schweifer, N.; Chiocca, S.; Decker, T.; et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol. Cell Biol. 2006, 26, 7913–7928. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [PubMed]
- Fulton, D.; Gratton, J.; McCabe, T.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.; Papapetropoulos, A.; Sessa, W. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [PubMed]
- Kawasaki, K.; Smith, R.S.; Hsieh, C.-M.; Sun, J.; Chao, J.; Liao, J.K. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol. Cell. Biol. 2003, 23, 5726–5737. [Google Scholar] [CrossRef] [PubMed]
- Rössig, L.; Li, H.; Fisslthaler, B.; Urbich, C.; Fleming, I.; Förstermann, U.; Zeiher, A.M.; Dimmeler, S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res. 2002, 91, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.-M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Kotchetkov, R.; Blaheta, R.; Driever, P.H.; Vogel, J.U.; Cinatl, J. Induction of differentiation and suppression of malignant phenotype of human neuroblastoma BE(2)-C cells by valproic acid: Enhancement by combination with interferon-α. Int. J. Oncol. 2002, 20, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Chelluri, R.; Caza, T.; Woodford, M.R.; Reeder, J.E.; Bratslavsky, G.; Byler, T. Valproic acid alters angiogenic and trophic gene expression in human prostate cancer models. Anticancer Res. 2016, 36, 5079–5086. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Suhan, T.; Michaelis, U.R.; Beek, K.; Rothweiler, F.; Tausch, L.; Werz, O.; Eikel, D.; Zörnig, M.; Nau, H.; et al. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. 2006, 13, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Wang, Y.W.; Chen, C.T.; Ho, C.M.; Su, W.H.; Jou, Y.S. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin. Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [PubMed]
- Balliu, M.; Guandalini, L.; Romanelli, M.N.; D’Amico, M.; Paoletti, F. HDAC-inhibitor (S)-8 disrupts HDAC6-PP1 complex prompting A375 melanoma cell growth arrest and apoptosis. J. Cell. Mol. Med. 2015, 19, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Woan, K.V.; Lienlaf, M.; Perez-Villaroel, P.; Lee, C.; Cheng, F.; Knox, T.; Woods, D.M.; Barrios, K.; Powers, J.; Sahakian, E.; et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 2015, 9, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Lienlaf, M.; Wang, H.-W.; Perez-Villarroel, P.; Lee, C.; Woan, K.; Rock-Klotz, J.; Sahakian, E.; Woods, D.; Pinilla-Ibarz, J.; et al. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J. Immunol. 2014, 193, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, M.; Gielen, P.; Brok, I.C.; Armandari, I.; Hoogerbrugge, P.M.; Adema, G.J. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014, 5, 6558–6572. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.R.; Malamas, A.S.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget 2016, 7, 7390–7402. [Google Scholar] [PubMed]
- Sabbatino, F.; Schwab, J.H.; Ferrone, S.; Ferrone, C.R. Evolution of studies of HLA class I antigen processing machinery (APM) components in malignant cells. Clin. Transpl. 2013, 1, 453–463. [Google Scholar]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Kortenhorst, M.S.; Wissing, M.D.; Rodríguez, R.; Kachhap, S.K.; Jans, J.J.; Van der Groep, P.; Verheul, H.M.; Gupta, A.; Aiyetan, P.O.; van der Wall, E.; et al. Analysis of the genomic response of human prostate cancer cells to histone deacetylase inhibitors. Epigenetics 2013, 8, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, A.; Ling, Q.-D.; Kumar, S.S.; Munusamy, M.A.; Alarfaj, A.A.; Chang, Y.; Kao, S.-H.; Lin, K.-C.; Wang, H.-C.; Umezawa, A. Generation of pluripotent stem cells without the use of genetic material. Lab. Investig. 2015, 95, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Burba, I.; Colombo, G.I.; Staszewsky, L.I.; De Simone, M.; Devanna, P.; Nanni, S.; Avitabile, D.; Molla, F.; Cosentino, S.; Russo, I.; et al. Histone deacetylase inhibition enhances self renewal and cardioprotection by human cord blood-derived CD34 cells. PLoS ONE 2011, 6, e22158. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Cen, J.; Li, J.; Zhao, R.; Zhu, C.; Wang, Z.; Xie, J.; Tang, W. Histone deacetylase inhibitor valproic acid (VPA) promotes the epithelial mesenchymal transition of colorectal cancer cells via up regulation of Snail. Cell Adhes. Migr. 2015, 9, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Luo, Z.; Yu, P.-J.; Xie, H.; He, Y.-W. Suberoylanilide hydroxamic acid (SAHA) promotes the epithelial mesenchymal transition of triple negative breast cancer cells via HDAC8/FOXA1 signals. Biol. Chem. 2016, 397, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Tabu, K.; Sasai, K.; Kimura, T.; Wang, L.; Aoyanagi, E.; Kohsaka, S.; Tanino, M.; Nishihara, H.; Tanaka, S. Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res. 2008, 18, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wan, H.; Xue, C.; Jiang, T.; Qian, C.; Zhang, Y. Histone deacetylase 3 implicated in the pathogenesis of children glioma by promoting glioma cell proliferation and migration. Brain Res. 2013, 1520, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.A.; Hraběta, J.; Groh, T.; Procházka, P.; Doktorová, H.; Eckschlager, T. Valproic acid increases CD133 positive cells that show low sensitivity to cytostatics in neuroblastoma. PLoS ONE 2016, 11, e0162916. [Google Scholar] [CrossRef] [PubMed]
- Rudà, R.; Pellerino, A.; Soffietti, R. Does valproic acid affect tumor growth and improve survival in glioblastomas? CNS Oncol. 2016, 5, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Hřebačková, J.; Poljaková, J.; Eckschlager, T.; Hraběta, J.; Procházka, P.; Smutný, S.; Stiborová, M. Histone deacetylase inhibitors valproate and trichostatin A are toxic to neuroblastoma cells and modulate cytochrome P450 1A1, 1B1 and 3A4 expression in these cells. Interdiscip. Toxicol. 2009, 2, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S. Histone deacetylases 1 and 2 regulate DNA replication and DNA repair: Potential targets for genome stability-mechanism-based therapeutics for a subset of cancers. Cell Cycle 2015, 14, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.C.; Jiang, S.; Zhou, X.C.; Hou, X.; Jin, F.; Podratz, K.C.; Jiang, S.-W. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol. Cancer Ther. 2006, 5, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.A.; Jones, A.J.; Avery, V.M.; Sumanadasa, S.D.M.; Ng, S.S.; Fairlie, D.P.; Adams, T.S.; Andrews, K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ezzeldin, H.H.; Diasio, R.B. Histone deacetylase inhibitors: Current status and overview of recent clinical trials. Drugs 2009, 69, 1911–1934. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Liao, W.S.L.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hoshino, K.; Sanchez-Gonzalez, B.; Kantarjian, H.; Garcia-Manero, G. Antileukemia activity of the combination of 5-aza-2′-deoxycytidine with valproic acid. Leuk. Res. 2005, 29, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Ecke, I.; Petry, F.; Rosenberger, A.; Tauber, S.; Mo, S.; Hess, I.; Dullin, C.; Kimmina, S.; Pirngruber, J.; Johnsen, S.A.; et al. Antitumor Effects of a Combined 5-Aza-2′ Deoxycytidine and Valproic Acid Treatment on Rhabdomyosarcoma and Medulloblastoma in Ptch Mutant Mice. Cancer Res. 2009, 69, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Klisovic, M.I.; Maghraby, E.A.; Parthun, M.R.; Guimond, M.; Sklenar, A.R.; Whitman, S.P.; Chan, K.K.; Murphy, T.; Anon, J.; Archer, K.J.; et al. Depsipeptide (FR 901228) promotes histone acetylation, gene transcription, apoptosis and its activity is enhanced by DNA methyltransferase inhibitors in AML1/ETO-positive leukemic cells. Leukemia 2003, 17, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.; Li, L.; Zhou, W.; Wu, L.; Zhao, Y.; Wang, D.; Lu, S.; Yu, Y.; Wang, H.; McNutt, M.A.; et al. HDAC inhibitors act with 5-aza-2′-deoxycytidine to inhibit cell proliferation by suppressing removal of incorporated abases in lung cancer cells. PLoS ONE 2008, 3, e2445. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, W.; Chen, G.; Zhang, H.; Jia, Y.; Wei, Y.; Yang, H.; Zhang, W.; Fiskus, W.; Bhalla, K.; et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate. Blood 2010, 116, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Greve, G.; Schiffmann, I.; Pfeifer, D.; Pantic, M.; Schüler, J.; Lübbert, M. The pan-HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR-mutated and -wildtype non-small cell lung cancer cells. BMC Cancer 2015, 15, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Takashina, T.; Kinoshita, I.; Kikuchi, J.; Shimizu, Y.; Sakakibara-Konishi, J.; Oizumi, S.; Nishimura, M.; Dosaka-Akita, H. Combined inhibition of EZH2 and histone deacetylases as a potential epigenetic therapy for non-small-cell lung cancer cells. Cancer Sci. 2016, 107, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.-Y.; Dai, Y.; Grant, S. Synergistic Induction of Oxidative Injury and Apoptosis in Human Multiple Myeloma Cells by the Proteasome Inhibitor Bortezomib and Histone Deacetylase Inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Andtbacka, R.H.I.; Dunner, K.; Pal, A.; Bornmann, W.G.; Chiao, P.J.; Huang, P.; et al. Aggresome disruption: A novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006, 66, 3773–3781. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, G.; Lembersky, D.; Kramer, L.; Fisher, R.I.; Friedberg, J.; Dent, P.; Grant, S. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood 2010, 115, 4478–4487. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, G.; Lembersky, D.; Son, M.P.; Attkisson, E.; Dent, P.; Fisher, R.I.; Friedberg, J.W.; Grant, S. Carfilzomib interacts synergistically with histone deacetylase inhibitors in mantle cell lymphoma cells in vitro and in vivo. Mol. Cancer Ther. 2011, 10, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Groh, T.; Hrabeta, J.; Khalil, M.A.; Doktorova, H.; Eckschlager, T.; Stiborova, M. The synergistic effects of DNA-damaging drugs cisplatin and etoposide with a histone deacetylase inhibitor valproate in high-risk neuroblastoma cells. Int. J. Oncol. 2015, 47, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Luchenko, V.L.; Salcido, C.D.; Zhang, Y.; Agama, K.; Komlodi-Pasztor, E.; Murphy, R.F.; Giaccone, G.; Pommier, Y.; Bates, S.E.; Varticovski, L. Schedule-dependent synergy of histone deacetylase inhibitors with DNA damaging agents in small cell lung cancer. Cell Cycle 2011, 10, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of Histone Deacetylase Increases Cytotoxicity to Anticancer Drugs Targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar] [PubMed]
- Catalano, M.G.; Poli, R.; Pugliese, M.; Fortunati, N.; Boccuzzi, G. Valproic acid enhances tubulin acetylation and apoptotic activity of paclitaxel on anaplastic thyroid cancer cell lines. Endocr. Relat. Cancer 2007, 14, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.H.; Yoon, W.S.; Park, K.Y.; Kim, S.M.; Lim, J.Y.; Woo, J.S.; Jeong, C.H.; Hou, Y.; Jeun, S.S. Valproic acid downregulates the expression of MGMT and sensitizes temozolomide-resistant glioma cells. J. Biomed. Biotechnol. 2012, 2012, 987495. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. DNA Methyltransferase and Histone Deacetylase Inhibitors in the Treatment of Myelodysplastic Syndromes. Semin. Hematol. 2008, 45, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Rudek, M.A.; Zhao, M.; He, P.; Hartke, C.; Gilbert, J.; Gore, S.D.; Carducci, M.A.; Baker, S.D. Pharmacokinetics of 5-azacitidine administered with phenylbutyrate in patients with refractory solid tumors or hematologic malignancies. J. Clin. Oncol. 2005, 23, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Badros, A.; Burger, A.M.; Philip, S.; Niesvizky, R.; Kolla, S.S.; Goloubeva, O.; Harris, C.; Zwiebel, J.; Wright, J.J.; Espinoza-Delgado, I.; et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin. Cancer Res. 2009, 15, 5250–5257. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Vesole, D.H.; Jagannath, S. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: A case series illustrating utility in clinical practice. Clin. Lymphoma Myeloma Leuk. 2010, 10, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Afifi, S.; Michael, A.; Azimi, M.; Rodriguez, M.; Lendvai, N.; Landgren, O. Role of Histone Deacetylase Inhibitors in Relapsed Refractory Multiple Myeloma: A Focus on Vorinostat and Panobinostat. Pharmacotherapy 2015, 35, 1173–1188. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Tambaro, F.P.; Bekele, N.B.; Yang, H.; Ravandi, F.; Jabbour, E.; Borthakur, G.; Kadia, T.M.; Konopleva, M.Y.; Faderl, S.; et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J. Clin. Oncol. 2012, 30, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Altman, J.K.; Licht, J.D. New strategies in acute myeloid leukemia: Redefining prognostic markers to guide therapy. Clin. Cancer Res. 2012, 18, 5163–5171. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIB Multicenter Trial of Vorinostat in Patients With Persistent, Progressive, or Treatment Refractory Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Marcucci, G. Targeting epigenetic changes in acute myeloid leukemia. Clin. Adv. Hematol. Oncol. 2005, 3, 855–865. [Google Scholar] [PubMed]
- Garcia-Manero, G.; Yang, H.; Bueso-Ramos, C.; Ferrajoli, A.; Cortes, J.; Wierda, W.G.; Faderl, S.; Koller, C.; Morris, G.; Rosner, G.; et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Campigotto, F.; Murphy, T.J.; Boswell, E.N.; Banwait, R.; Azab, F.; Chuma, S.; Kunsman, J.; Donovan, A.; Masood, F.; et al. Results of a phase 2 trial of the single-agent histone deacetylase inhibitor panobinostat in patients with relapsed/refractory Waldenstrom macroglobulinemia. Blood 2013, 121, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.A.; Advani, A.; Fernandez, L.; Van Der Jagt, R.; Brandwein, J.; Kambhampati, S.; Kassis, J.; Davis, M.; Bonfils, C.; Dubay, M.; et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br. J. Haematol. 2009, 147, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009, 114, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.; Fischer, T.; Cortes, J.; Garcia-Manero, G.; Beck, J.; Ravandi, F.; Masson, E.; Rae, P.; Laird, G.; Sharma, S.; et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin. Cancer Res. 2006, 12, 4628–4635. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Futur. Med Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- O'connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 2006, 24, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Gorlia, T.; Cairncross, J.G.; Van Den Bent, M.J.; Mason, W.; Belanger, K.; Brandes, A.A.; Bogdahn, U.; Macdonald, D.R.; Forsyth, P.; et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 2011, 77, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Scherpereel, A.; Berghmans, T.; Lafitte, J.J.; Colinet, B.; Richez, M.; Bonduelle, Y.; Meert, A.P.; Dhalluin, X.; Leclercq, N.; Paesmans, M.; et al. Valproate-doxorubicin: Promising therapy for progressing mesothelioma. A phase II study. Eur. Respir. J. 2011, 37, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Jotte, R.M.; Konduri, K.; Neubauer, M.A.; Spira, A.I.; Ruxer, R.L.; Varella-Garcia, M.; Bunn, P.A.; Hirsch, F.R. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 2012, 30, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic therapeutics: A new weapon in the war against cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mechanism of anticancer effects of HDAC inhibitors. ROS: reactive oxygen species.


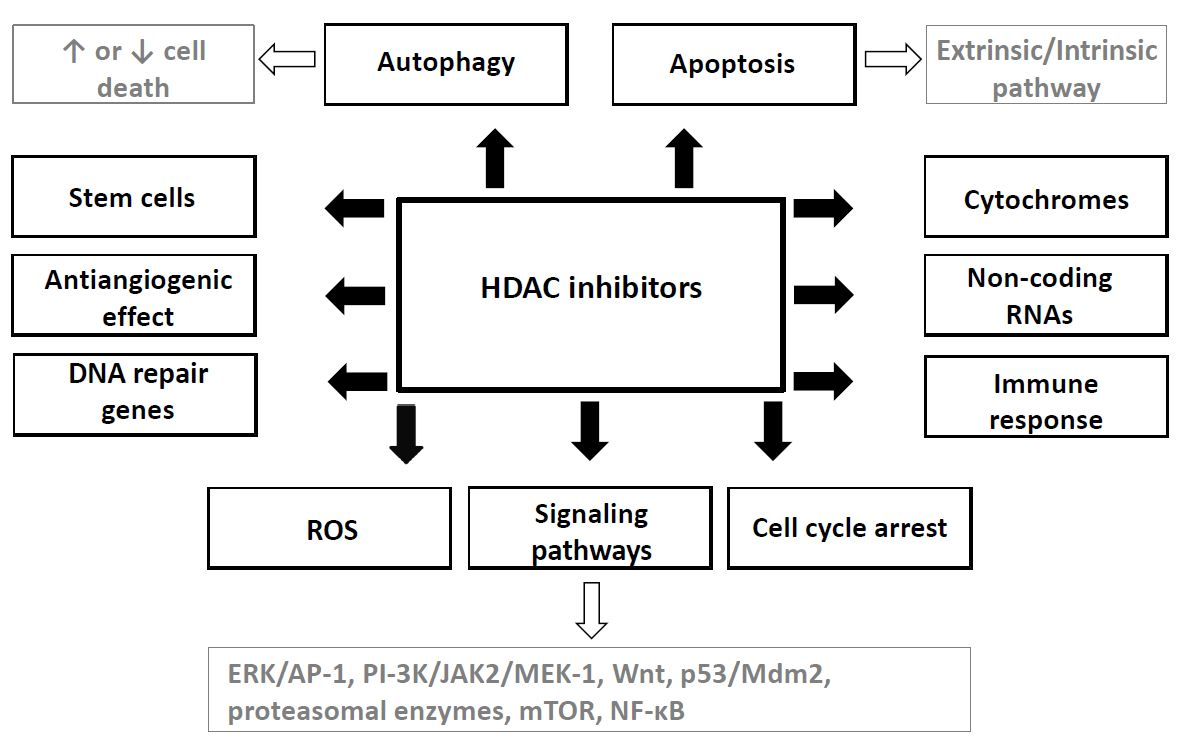{kind=link}
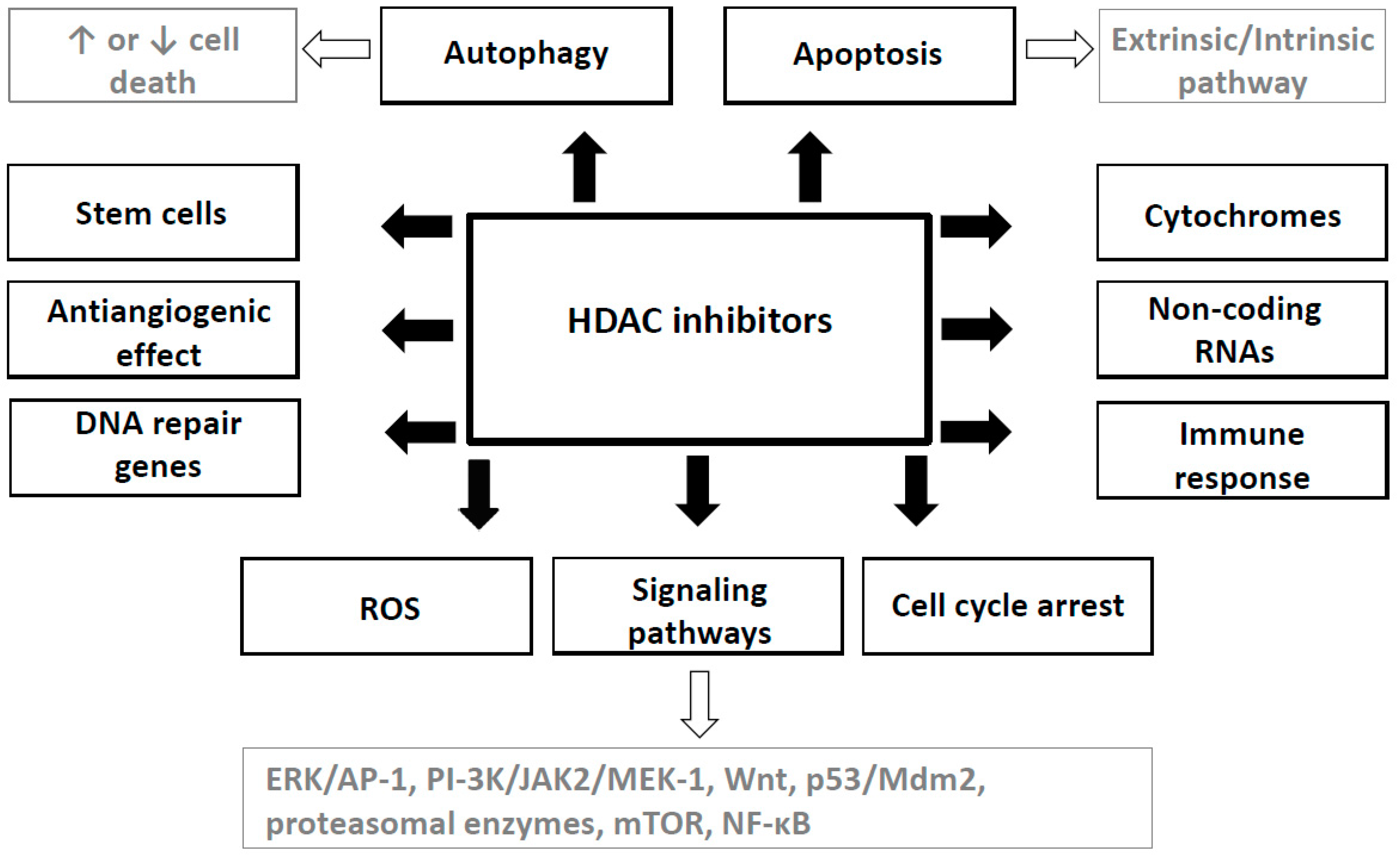{kind=link}
Table 1.
Overview of selected histone deacetylases (HDAC) inhibitors.
| Class | HDAC Inhibitor | Target HDAC Class | Clinical Status |
|---|---|---|---|
| hydroxamic acids | Trichostatin A | pan | preclinical |
| SAHA | pan | approved for cutaneous T-cell lymphoma | |
| Belinostat | pan | approved for peripheral T-cell lymphoma | |
| Panabiostat | pan | approved for multiple myeloma | |
| Givinostat | pan | phase II clinical trials—relapsed leukemia and multiple myeloma | |
| Resminostat | pan | phase I and II clinical trials—hepatocellular carcinoma | |
| Abexinostat | pan | phase II clinical trial—B-cell lymphoma | |
| Quisinostat | pan | phase I clinical trial—multiple myeloma | |
| Rocilinostat | II | phase I clinical trial—multiple myeloma | |
| Practinostat | I, II and IV | phase II clinical trial—prostate cancer | |
| CHR-3996 | I | phase I clinical trial—advanced/metastatic solid tumors refractory to standard therapy | |
| short chain fatty acids | Valproic acid | I, IIa | approved for epilepsia, bipolar disorders and migraine, phase II clinical trials—several studies |
| Butyric acid | I, II | phase II clinical trials—several studies | |
| Phenylbutyric acid | I, II | phase I clinical trials—several studies | |
| benzamides | Entinostat | I | phase II clinical trials—breast cancer, Hodgkin´s lymphoma, non-small cell lung cancer, phase III clinical trial—hormone receptor positive breast cancer |
| Tacedinaline | I | phase III clinical trial—non-small cell lung cancer and pancreatic cancer | |
| 4SC202 | I | phase I clinical trial—advanced hematological malignancies | |
| Mocetinostat | I, IV | phase II clinical trials—Hodgkin´s lymphoma | |
| cyclic tetrapeptides | Romidepsin | I | approved for cutaneous T-cell lymphoma |
| sirtuins inhibitors | Nicotinamide | all class III | phase III clinical trial—laryngeal cancer |
| Sirtinol | SIRT 1 and 2 | Preclinical | |
| Cambinol | SIRT 1 and 2 | Preclinical | |
| EX-527 | SIRT 1 and 2 | cancer preclinical, phase I and II clinical trials—Huntington disease, glaucoma |
Table 2.
Main mechanism of anticancer effects of HDAC inhibitors—preclinical studies.
| Mechanism | Target | Model | HDAC Inhibitor | Ref. |
|---|---|---|---|---|
| Cell cycle arrest | CDKN1A/p21 | in vitro leukemic cells U937 | SAHA | [42] |
| CDKN1A/p21 | in vitro bladder cancer cells T24 | SAHA | [43] | |
| CDKN1A/p21 | in vitro normal breast MCF-10A, prostate cancer PC-3, DU145, colon cancer SW620, ovarian cancer IGROV, breast cancer MCF-7, lung cancer A549 | FR901228 | [44] | |
| p53 | in vitro colon cancer HCT116 + knock out DNMT1−/−, DNMT3B−/−, DNMT1−/− and DNMT3B−/− HCT116 | TSA | [46] | |
| p53 | in vitro lung cancer A549 | depsipeptide | [47] | |
| p53 | in vitro lung cancer H1299, osteosarcoma U2OS, human embryonal kidney HEK293 | TSA, nicotinamide | [48] | |
| p21 | in vitro colon cancer HCT116 + knock out p53−/−, p21−/− | sodium butyrate | [49] | |
| p21 | in vitro gastric cancer TMK-1, MKN-1, MKN-7, MKN-28, MKN-74, MKN-45, KATO III, HSC-39, oral squamous carcinoma HSC-4, Ho-1-N-1, Ho-1-U-1 | TSA | [50] | |
| RUNX3 | in vitro pancreatic endocrine tumors- insulinoma CM, carcinoid BON, somatostatinoma QCP-1 | TSA | [52] | |
| RUNX3 | in vitro prostate cancer DU-145, LNCaP, PC-3 | TSA | [55] | |
| Apoptosis | death receptor | in vitro leukemic cells HL60 | apicidin | [60] |
| death receptor | in vitro AML cells from patients, normal CD34+ progenitors | MS275 | [61] | |
| intrinsic pathway | in vitro leukemic cells U937 | SAHA | [42] | |
| intrinsic pathway | in vitro T cell leukemia CEM-CCRF and doxorubicin derived P-gp+(CEM-P-gp) | SAHA | [63] | |
| intrinsic pathway | in vitro fetal lung fibroblasts IMR90, osteosarcoma U2OS and Saos-2, colon cancer HCT116 p53−/− | SAHA, TSA | [64] | |
| intrinsic pathway | in vitro leukemic cells Jurkat, ML-1 | MS275 | [66] | |
| intrinsic pathway | in vitro leukemic cells Jurkat, U937, HL-60, K562, U937 expressing Bcl-2, Bcl-XL, CrmA, C8DN, p21 antisense | MS275 | [67] | |
| intrinsic pathway | in vitro prostate cancer LNCaP, bladder cancer T24, breast cancer MCF7 | SAHA | [68] | |
| intrinsic pathway | in vitro neuroblastoma UKF-NB-4, normoxic and hypoxic condition | VPA | [71] | |
| Autophagy | FOXO3 | in vitro 293T, mice embryonal fibroblasts wild t. and SIRT−/−, Rat1 fibroblasts inducible expressing FOXO3 | Nicotinamide, BML-210, splitomicin, TSA | [74] |
| Atg5 & Beclin1 | in vitro rat cardiomyocytes, in vivo model- a-MHC Beclin transgenic mice | TSA | [75] | |
| unknown | in vitro cervical cancer HeLa | siRNA HDAC1, FK228, SAHA | [76] | |
| Atg4D | in vitro neuroblastoma BE(2)-C, Kelly, IMR32 | siRNA HDAC10 | [77] | |
| Atg5, 7 & 8 | in vitro cervical cancer HeLa, colon cancer HCT116- SIRT1 transfected, starvation | - | [78] | |
| FOXO1 | in vitro colon cancer HCT116 Tp53−/− and +/+, mouse embryonic fibroblasts, liver cancer HepG2 | TSA | [79] | |
| Akt/mTOR, ULK1 | in vitro liver cancer Hep3B, HepG2, Huh7, mouse embryonic fibroblasts | SAHA, TSA, MS275 | [80] | |
| mTOR | in vitro endometrial stromal sarcoma ESS-1, endometrial stromal cells HESCs | SAHA | [81] | |
| p53 | in vitro uterine sarcoma MES-SA, ESS-1, cervical cancer HeLa, PANC-1, leukemic cells, pancreatic cancer Jurkat, HL-60, U937 | SAHA | [82] | |
| ULK1 | in vitro glioblastoma T98G, MEF cells, ULK1/2 knockout MEF, ATG3 knockout MEFs | SAHA | [83] | |
| ULK1 | in vitro leukemic cells Jurkat | SAHA | [84] | |
| NF- κB | in vitro PC3, DU145, and HCT116, mouse embryonic fibroblasts and MEF Atg5−/− | SAHA, MS275 | [85] | |
| Akt/mTOR | short term culture glioblastoma cells xenografts in nude mice | SAHA | [87] | |
| Effect on non-coding RNA | miR-129-5p → GALNT1 & SOX4 | in vitro thyroid carcinoma BCPAP, TPC-1, 8505C, CAL62 | TSA, SAHA | [88] |
| miR-31→BMI1 | in vitro breast cancer MDA-MB-231, MCF7, 293T, embryonal lung fibroblasts | sodium butyrate, panobiostat | [89] | |
| miR-15 & let-7 → Myc | in vitro and mice xenografts leukemic cells/lymphoma Daudi, Ramos, Raji, Su-DHL-6, NIH3T3, P493-6 | RGFP966, depsipetide | [90] | |
| Drosha/DGCR8 | in vitro 293FT embryonal kidney cells HDAC1 transfected | / | [91] | |
| miR-449a → HDAC1 | in vitro prostate cancer PC-3 | siRNA HDAC1 | [92] | |
| uc002mbe.2 lncRNA | in vitro liver cancer Huh7, Bel7402, Bel7721, and HepG2, ShRNA uc002mbe.2 lncRNA | TSA | [95] | |
| lncRNA Xist | in vitro stem cells from 16 breast cancer cell lines | SAHA, VPA, abexinostat | [97] | |
| Effect on signal pathways | c-Jun | in vitro neuroblastoma SH-SY5Y | VPA | [98] |
| ERK | in vitro neuroblastoma SH-SY5Y | VPA | [65] | |
| APC/β-catenin/c-Myc | in vitro colon cancer HT-29 HDAC2 transfected, mice C57BL/6J-APC-/- | VPA | [10] | |
| GSK3 β | in vitro hematopoetic stem cells | VPA | [104] | |
| ubiquitin–proteasome | in vitro embryonal kidney cells HEK293T | VPA, TSA | [105] | |
| Anti-angiogenic effect | Gja1, Irf1, Gbp2 | in vitro embryonic stem cells HDAC1 mutated and wild type | / | [106] |
| ACNA2D2 | HDAC1 or HDAC2 mutated C57BL/6 mice | / | [107] | |
| PPAR, ERR | HDAC3 conditional C57BL/6 mice | / | [108] | |
| eNOS, Akt | in vitro endothelial cells, Akt transfected, mutatnteNOS | / | [110] | |
| eNOS | in vitro HUVEC | TSA, MS275 | [112] | |
| MMP-2/VEGF | in vitro melanoma A375, toxicity tests mice | Compound 8 | [119] | |
| semaphoring III | in vitro HUVEC | TSA, SAHA | [113] | |
| thrombospondin-1, activin | in vitro, neuroblastoma BE(2)-C | VPA | [114] | |
| Modulation of immune response | TAP1 & 2, LMP-2, tapasin | in vitro+mice xenografts mouse lung TC-1, rat pancreas D11, mouse fibroblasts A9, endothelial cells PA, mouse colinic cells LMD, mouse melanoma B16F10, B16F10/TAP-1 | TSA | [118] |
| MHC class I | mice C57BL and B6.CB17-Prkdc, human melanoma samples, mouse melanoma B16-F10-luc | MGCD0103, LBH589, TSA | [120] | |
| STAT3/IL-10 | BALB/c and C57BL/6 mice, TCR transgenic mice, HDAC6 recombinant mutants | / | [121] | |
| Tumor associated antigens | in vitro prostate cancer LNCaP, breast cancer MDA-MB-231 | SAHA, entinostat | [123] | |
| MHC genes | in vitro prostate cancer PCa and DU-145 | VPA | [126] |
Table 3.
Combination of HDAC inhibitors with other anticancer therapy.
| HDAC Inhibitor | Other Anticancer Therapy | Model | Effect | Ref. |
|---|---|---|---|---|
| SAHA, tubacin | etoposide, doxorubicin | in vitro prostate cancer LNCaP, breast cancer MCF-7, normal human foreskin fibroblast cells | Potentiation in ca cells not in fibroblasts | [140] |
| TSA | 5-aza-2′-deoxycytidine | in vitro pancreatic endocrine cancer cells | Potentiation | [52] |
| TSA | 5-aza-2′-deoxycytidine | in vitro LNCaP, DU-145, and PC-3 prostate cancer | Potentiation | [55] |
| SAHA, TSA | 5-aza-2′-deoxycytidine | in vitro ovarian cancer Hey, SKOv3 | Synergism | [142] |
| VPA | 5-aza-2′-deoxycytidine | in vitro leukemic cells HL-60, MOLT4 | Synergism | [143] |
| VPA | 5-aza-2′-deoxycytidine | Ptch knockout mice | Potentiation | [144] |
| Depsipeptide | 5-aza-2′-deoxycytidine | in vitro Kasumi-1 cells and blasts from patient with t(8;21) AML | Potentiation | [145] |
| TSA | 5-aza-2′-deoxycytidine | in vitro lung cancer A549, H719 | Synergism | [146] |
| SAHA | β-phenylethyl isothiocyanate | in vitro leukemia cells HL60, HL60/LR, HL60/C6F, U937, ML1, samples from patients with acute myeloid leukemia | Potentiation | [147] |
| Panobinostat | erlotinib | in vitro lung cancer HCC827, A549, NCI-H460 (EGFR wild type and mutant) | Synergism/Potentiation | [148] |
| SAHA | 3-deazaneplanocin A (EZH2 inhibitor) | in vitro and BALB/cAJcl-nu/nu mice xenografts lung cancer NCI-H1299, NCI-H1975, A549, PC-3 | Synergism | [149] |
| sodium butyrate, SAHA | bortezomib | in vitro multiple myeloma U266, RPMI8226 and patient samples | Synergism | [150] |
| SAHA | bortezomib | in vitro and nude mice xenografts, pancreatic cancer L3.6pl, pancreatic epitelium HPDE6-E6E7 | Potentiation | [151] |
| SAHA | carfilzomib | in vitro and nude mice xenografts, lymphoma cell SUDHL16, SUDHL4 , SUDHL6, OCI-LY10, OCI-LY3 bortezomib-resistant SUDHL16-10BR, OCI-LY10-40BR and lymphoma cells frompatients | Potentiation | [152] |
| SNDX-275, SAHA | carfilzomib, bortezomib | in vitro and nude mice xenografts, lymphoma cell Granta 519, Rec-1, HF-4B, JVM-2, MINO, JVM-13 | Potentiation | [153] |
| VPA | cisplatin, etoposide | in vitro neuroblastoma cell UKF-NB-4 | Potentiation-schedule dependent | [154] |
| belinostat, romidepsin | cisplatin, etoposide | in vitro lung cancer H82, H146, H526 and H446 | Synergism-schedule dependent | [155] |
| TSA, SAHA | VP-16, ellipticine, 5fluorouracil, doxorubicin, cisplatin, cyclophosphamide, campthotecin | in vitro glioblastoma U118, brest cancer MCF-7, normal breast MCF-12F, normal intestinal epithelia FHs74Int | Potentiation-schedule dependent, except of 5fluorouracil, cyclophosphamide, campthotecin | [156] |
| VPA | paclitaxel | in vitro anaplastic thyroid carcinoma CAL-62, ARO | Potentiation | [157] |
| VPA | Gene therapy (MSC + HSV-TK) | glioblastoma U87 xenografts in nude mice | Potentiation | [158] |
MSC + HSV-TK- herpes simplex thymidine kinase transfected bone marrow mesenchymal stem cells.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. https://doi.org/10.3390/ijms18071414
AMA Style
Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. International Journal of Molecular Sciences. 2017; 18(7):1414. https://doi.org/10.3390/ijms18071414
Chicago/Turabian StyleEckschlager, Tomas, Johana Plch, Marie Stiborova, and Jan Hrabeta. 2017. "Histone Deacetylase Inhibitors as Anticancer Drugs" International Journal of Molecular Sciences 18, no. 7: 1414. https://doi.org/10.3390/ijms18071414
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.