
Abstract
The tumour suppressor gene TP53 is mutated in ~50% of human cancers. In addition to its function in tumour suppression, p53 also plays a major role in the response of malignant as well as nontransformed cells to many anticancer therapeutics, particularly those that cause DNA damage. P53 forms a homotetrameric transcription factor that is reported to directly regulate ~500 target genes, thereby controlling a broad range of cellular processes, including cell cycle arrest, cell senescence, DNA repair, metabolic adaptation and cell death. For a long time, induction of apoptotic death in nascent neoplastic cells was regarded as the principal mechanism by which p53 prevents tumour development. This concept has, however, recently been challenged by the findings that in striking contrast to Trp53-deficient mice, gene-targeted mice that lack the critical effectors of p53-induced apoptosis do not develop tumours spontaneously. Remarkably, even mice lacking all mediators critical for p53-induced apoptosis, G1/S boundary cell cycle arrest and cell senescence do not develop any tumours spontaneously. In this review we discuss current understanding of the mechanisms by which p53 induces cell death and how this affects p53-mediated tumour suppression and the response of malignant cells to anticancer therapy.
Similar content being viewed by others

Main

Facts
-
TP53 is a critical tumour suppressor that is mutated in ~50% of human cancers.
-
In unstressed cells p53 protein levels are very low because it is targeted for proteasomal degradation by the E3 ubiquitin ligase MDM2.
-
TP53 is activated in response to many stress stimuli, including activation of oncogenes and DNA damage.
-
Upon activation, p53 directly regulates the transcription of ~500 genes and indirectly regulates many additional genes and thereby controls diverse cellular processes.
-
P53 induces apoptosis in nontransformed cells mostly by direct transcriptional activation of the pro-apoptotic BH3-only proteins PUMA and (to a lesser extent) NOXA.
-
Combined loss of the p53 effectors of apoptosis (PUMA plus NOXA) and cell cycle arrest/cell senescence (p21) does not cause spontaneous tumour development.
-
Apoptosis induction via PUMA and NOXA is critical for the killing of malignant cells by anticancer drugs that activate TP53 but other effectors contribute also.
Open questions
-
Which processes and target genes activated by p53 are critical for the prevention of cancer?
-
Loss of which p53-induced processes cooperate with loss of p53-induced apoptosis to cause cancer?
-
Why do certain malignant as well as nontransformed cells undergo apoptosis upon TP53 activation, whereas others do not die, but instead undergo cell cycle arrest and/or senescence?
-
What are the differences in p53-induced apoptosis between nontransformed and malignant cells?
-
How do the hot spot p53 mutant proteins inhibit wild-type p53-induced apoptosis in nascent neoplastic as well as malignant cells?
Discovery of p53 and Discovery of Mutations in the TP53 Gene in Human Cancer
The p53 protein (also called TP53) was discovered as a protein bound to the SV40 large T antigen in transformed cells (reviewed in Levine et al.1 and Lane and Benchimol2). Inadvertently, some of the first experiments in which p53 was overexpressed in cell lines used constructs encoding cancer-derived mutant TP53. Such enforced mutant p53 expression enhanced cell growth, and it was therefore concluded that p53 functions as an oncoprotein (reviewed in Levine et al.1 and Lane and Benchimol2). Subsequent studies found, however, that enforced expression of wild-type (WT) TP53 actually impaired the growth of transformed cells in culture, providing the first evidence that TP53 can function as a tumour suppressor.3, 4 At about the same time, it was discovered that many sporadic human cancers of diverse origins carried mutations in TP53, usually a point mutation in one allele that permitted expression of mutant p53 protein (often at abnormally high levels; see below) accompanied by a deletion that removed the other allele, including adjoining regions.5, 6 Moreover, individuals with the Li–Fraumeni syndrome, who carry germline heterozygous mutations in TP53, usually develop multiple cancers over their lifetime, often from a young age.7, 8
Most of the mutations in TP53 detected in cancer cells are point mutations in the DNA-binding domain. These mutant p53 proteins are thought to be unable to regulate the transcription of WT p53 target genes (loss of function (LOF)) (reviewed in Vousden and Lane9 and Freed-Pastor and Prives10). Interestingly, many mutant p53 proteins are detected at high levels in malignant cells. Therefore, by forming mixed tetramers with WT p53, mutant p53 proteins can exert dominant negative effects (DNEs) that are likely to play critical roles early during transformation when nascent neoplastic cells still retain their WT TP53 allele (reviewed in Vousden and Lane9 and Freed-Pastor and Prives10). In addition, certain p53 mutants have been reported to exert gain-of-function (GOF) effects by binding to and thereby modulating the functions of other tumour suppressors and transcriptional regulators (reviewed in Vousden and Lane9 and Freed-Pastor and Prives10). It remains unclear which of the LOF, the DNE or the GOF effects of mutant p53 are most important during the development and sustained growth of a cancer, and it appears likely that this may vary depending on both the cell of origin undergoing transformation and the nature of the cooperating oncogenic lesions that drive the neoplastic transformation of these cells.
Control of TP53 Activation and Cellular Responses Activated by p53
Unstressed, nontransformed cells contain very low (often undetectable) levels of WT p53 protein despite readily detectable mRNA expression.11 The main reason for this is that p53 is targeted for proteasomal degradation by the E3 ligase, MDM2 (Figure 1).12, 13, 14 In response to diverse stress stimuli, including activation of oncogenes, DNA damage or nutrient deprivation, the levels of p53 protein rise substantially because several signalling pathways that are activated in response to the aforementioned stressors converge upon the inhibition of MDM2, whereas some lead to modifications (e.g., acetylation, phosphorylation) in the p53 protein itself (Figure 1) (see reviews9, 10, 15). Upon activation, p53 binds as a homotetramer to specific sequences in the regulatory regions of its target genes (~500).16, 17, 18, 19, 20 Studies using enforced expression or conditional activation (e.g., using temperature-sensitive mutants) of p53 in cell lines revealed that p53 can activate diverse cellular effector processes, including cell cycle arrest, cellular senescence, coordination of various DNA damage repair pathways, metabolic adaptation and apoptotic cell death (reviewed in Vousden and Lane9 and Freed-Pastor and Prives10). Gene expression studies and functional assays using gene-targeted mice soon identified genes that are essential for certain p53-activated cellular responses. For example, the cyclin-dependent kinase inhibitor (CDKi) p21 is critical for p53-mediated G1/S boundary cell cycle arrest and cell senescence21 (although additional p53 target genes also play a role in the latter process). Moreover, several genes implicated in various DNA repair processes were found to be either direct targets of p53 or indirectly regulated by p53.22 Finally, direct transcriptional induction of Mdm2 by p53 was recognised as a major negative feedback loop in p53 signalling.23 This is most spectacularly demonstrated by the finding that loss of MDM2 causes excess p53 activation, resulting in early embryonic lethality in mice, and that this lethality can be prevented by concomitant loss of Trp53.24, 25

Regulation of p53 protein level and activity in unstressed versus stressed cells. Models depicting the mechanisms that regulate p53 protein levels and activity in unstressed cells and in cells undergoing stress, for example, due to the activation of oncogenes or DNA lesions that they have sustained. (Ub, ubiquitin; P, phosphorylation; Ac, acetylation)
P53-Mediated Induction of Apoptosis
The first clue that p53 can induce apoptotic cell death came from studies using a myeloid leukaemia cell line expressing a temperature-sensitive conditionally active mutant of p53 (i.e., at 37 °C this protein behaves as mutant p53 but at 32 °C it assumes WT p53 structure and function).26 The observation that p53 can induce apoptosis was confirmed and extended by similar experiments in which a temperature-sensitive p53 or WT p53 was also enforcibly expressed in erythroleukaemia cells,27 a colon cancer cell line28 and a Burkitt lymphoma line.29
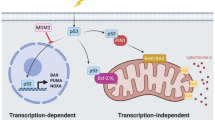
There are two distinct, although ultimately converging, pathways to apoptosis in mammalian cells:30 the so-called BCL-2-regulated (also called intrinsic, mitochondrial or stress) pathway that is activated by stress conditions, such as cytokine deprivation, ER stress or DNA damage, and the so-called death receptor (also called extrinsic) pathway that is activated by ligation of members of the tumour necrosis factor receptor (TNFR) family bearing an intracellular death domain.31, 32, 33 In the BCL-2-regulated apoptotic pathway, cell death is initiated by the transcriptional and/or post-transcriptional upregulation of the so-called pro-apoptotic BH3-only members of the BCL-2 protein family (BIM, PUMA, BID, BMF, BAD, BIK, NOXA, HRK). The BH3-only proteins bind and inhibit the pro-survival BCL-2 proteins (BCL-2, BCL-XL, MCL-1, BCL-W and A1/BFL1), thereby unleashing the cell death effectors BAX and BAK (the pro-apoptotic multi-BH domain members of the BCL-2 family that may also include BOK34, 35, 36, 37). Certain BH3-only proteins were reported to also activate BAX/BAK directly (see reviews32, 38). Activation of BAX/BAK causes mitochondrial outer membrane permeabilisation (MOMP), the point of no return in apoptosis signalling, with consequent activation of the cascade of aspartate-specific cysteine proteases (caspases; in this pathway initiated by caspase-939, 40, 41 and its activator APAF-142, 43) that dismantle the cell (Figure 2) (reviewed in Green32). Conversely, the death receptor pathway activates apoptosis by recruitment and activation of the pro-form of caspase-8 via the adaptors FADD, and in some cases also TRADD, at the ligated death receptors at the plasma membrane.44, 45 In so-called type 1 cells (e.g., thymocytes), such caspase-8 activation with consequent activation of the effector caspases (caspase-3 and -7) is sufficient for effective induction of apoptosis. In contrast, in the so-called type 2 cells (e.g., hepatocytes), efficient cell killing requires amplification of the caspase cascade by crossover activation of the BCL-2-regulated apoptotic pathway that is achieved by caspase-8-mediated proteolytic activation of the otherwise inert BH3-only protein BID.46, 47, 48, 49, 50

Mechanisms of p53-induced apoptosis. Model depicting the mechanism by which activated p53 induces apoptosis through the BCL-2-regulated pathway. Fat arrows indicate p53-induced targets that are essential for p53-induced apoptosis. Thin arrows indicate p53-induced targets that are constituents of the BCL-2-regulated apoptotic pathway but are still expressed at levels sufficient for apoptosis induction in the complete absence of p53; that is, their induction by p53 may make the pathway work more efficiently, but this induction is not a sine qua non for p53-induced apoptosis, at least in haematopoietic cells. The broken arrow indicates that p53 may also activate BIM expression indirectly. The possible scenario that activation of targets that are not constituents of the apoptosis machinery per se can impact on apoptosis indirectly is also depicted
Studies using cell lines with enforced expression of WT p53 or temperature-sensitive p53 revealed that overexpression of anti-apoptotic BCL-2 could prevent p53-induced apoptosis.51, 52, 53 Notably, the cells rescued from p53-induced death by expression of BCL-2 still underwent cell cycle arrest,52 demonstrating that p53 was fully functional (i.e., BCL-2 does not directly block all p53 functions). Thus, p53 must induce cell cycle arrest and apoptosis through distinct pathways, and BCL-2 (or other pro-survival BCL-2 family members) inhibit p53-induced apoptosis at a downstream point in apoptosis signalling (Figure 2).
The caveat with the aforementioned experiments is that the levels of p53 used to induce apoptosis were abnormally high. Hence, it was not yet proven that p53 could induce apoptosis under physiological conditions, that is, when expressed at normal levels. This was established when it was shown that thymocytes and other lymphoid cell subsets from Trp53 knockout mice are completely resistant to apoptosis induced by γ-radiation and treatment with chemotherapeutic drugs that induce DNA damage (e.g., etoposide, cyclophosphamide, cisplatin).52, 54, 55
Discovery of the p53-Activated Inducers of the BCL-2-Regulated Apoptotic Pathway
The demonstration that p53-induced apoptosis can be blocked by BCL-2 overexpression launched the hunt to identify the p53-activated initiators of the cell death pathway that is regulated by BCL-2. Many candidates were identified by searching for genes that were upregulated in response to overexpression of p53 at highly nonphysiological levels. Perhaps predictably, most of these candidates have still not been proven to have roles in apoptosis. The two notable exceptions are Puma/Bbc3 and Noxa/Pmaip: both of these genes are direct p53 targets and encode pro-apoptotic BH3-only proteins.56, 57, 58, 59 These genes are directly upregulated by p53 and their enforced expression causes rapid apoptosis in cell lines, whereas their knockdown protects cells against cytotoxic stimuli that trigger apoptosis in a p53-dependent manner.56, 57, 58, 59 Studies with gene-targeted mice revealed that PUMA and to a lesser extent NOXA are critical for p53-mediated apoptosis (e.g., apoptosis induced by γ-radiation or chemotherapeutic drugs that cause DNA damage) in a broad range of cell types, including lymphoid as well as myeloid cells, fibroblasts and skin keratinocytes, both in culture and in vivo.60, 61, 62, 63 Remarkably, thymocytes from Puma/Noxa double knockout mice are as resistant to γ-radiation in vivo as those from Trp53 knockout mice (Figure 2).64 Loss of PUMA generally affords many cell types with much greater protection against cytotoxic agents that trigger apoptosis via p53 activation (e.g., γ-radiation in lymphoid cells) than loss of NOXA.61, 64 Curiously, however, in certain cell types and under certain conditions the impact of loss of NOXA is more pronounced. For example, NOXA deficiency protects skin keratinocytes and fibroblasts more potently against UV radiation (a p53-dependent apoptotic stimulus) than loss of PUMA.63 This suggests that the relative contributions of PUMA versus NOXA to the induction of apoptosis may vary depending on the cytotoxic insult, the nature of the responding cell or both. Of note, NOXA preferentially inhibits MCL-1 and (in contrast to other BH3-only proteins that can bind to MCL-1) promotes the degradation of this pro-survival protein.65, 66 The prominent role of NOXA in UV radiation-induced apoptosis may therefore be explained if MCL-1 is the critical pro-survival protein protecting skin keratinocytes and fibroblasts against UV radiation.
Even though in non-transformed cells combined loss of PUMA and NOXA provides full protection (i.e., as potent as loss of TP53 itself) against apoptosis induced by γ-radiation or chemotherapeutic drugs that induce DNA damage,64 this is not the case in malignant lymphoma and leukaemia cells. For example, mouse Eμ-Myc lymphoma cells lacking both PUMA and NOXA are much less resistant to cyclophosphamide, etoposide or nutlin-3a (that activates p53 in a nongenotoxic manner by blocking its major inhibitor, the MDM2 E3 ubiquitin ligase67) than loss of p53 or overexpression of anti-apoptotic BCL-2.68, 69 Interestingly, additional loss of the BH3-only protein BIM70, 71 (i.e., combined loss of PUMA, NOXA plus BIM) provided as potent protection against nutlin-3a and etoposide as loss of p53 (Figure 2).68, 69 After treatment with etoposide or nutlin-3a, BIM expression was upregulated in Eμ-Myc lymphoma cells at a considerably later time compared with the induction of PUMA and NOXA.68, 69 Therefore, p53 may indirectly regulate BIM expression, perhaps through repression of a microRNA that regulates BIM. However, evidence for direct activation of Bim transcription by p53 has also been reported.72, 73, 74 Collectively, these findings reveal that p53-induced apoptosis is likely to be more complex in malignant cancer cells compared with nontransformed cells (Figure 2).
It is also noteworthy that two additional constituents of the BCL-2-regulated apoptotic pathway, the pro-apoptotic effector BAX and APAF-1 (the scaffold protein for caspase-9 activation) have been convincingly shown to be transcriptionally regulated by p53.75, 76, 77 However, p53 is not a sine qua non for BAX and APAF-1 expression. This is best demonstrated by the observations that Trp53-deficient haematopoietic cells express normal levels of BAX and APAF-1 and undergo apoptosis as readily as control (wild-type) cells after exposure to cytotoxic insults that induce BAX/BAK-dependent apoptosis,78 involving APAF-1,42, 43, 79 in a p53-independent manner (e.g., cytokine deprivation, treatment with glucocorticoids).52 Accordingly, we conclude that p53-mediated transcriptional induction of BAX and APAF-1 is not essential for induction of apoptosis, at least in haematopoietic cells, but may serve in certain other cell types, as a mechanism to make the system work more efficiently or even allow this pathway to operate. This may relate to the observation that the levels of BAX, APAF-1 and other constituents of the apoptosis machinery are much lower in many tissues (e.g., heart, kidney, brain) in adult mice and humans compared with newborns. This may account for the reduced sensitivity to apoptotic stimuli of cells from these tissues in adults compared with newborns.80
Mechanisms of Induction of Apoptosis by p53-Related Proteins
For many years p53 was thought to have no relatives, but then within a short time frame, two closely related proteins, called p6381 and p73,82 were discovered. P63 and p73 share similarity with p53 in their DNA-binding and transactivation domains and it is therefore widely assumed that many recognised p53 target genes, and hence the cellular processes they control, can also be regulated by p63 and p73.83 Some early studies showed that overexpression (although at clearly nonphysiological levels) of p63 or p73 can cause cell death with morphological and biochemical features of apoptosis.81, 84 The first demonstration that p63 can induce apoptosis under physiological conditions came from elegant studies in the mouse ovary. Even very low dose (0.5 Gy) γ-radiation kills all primordial follicles in 5-day-old female mice and renders these animals permanently infertile. This cell death is completely prevented by loss of p63, but loss of p53 has no protective effect.85 Combined loss of PUMA and NOXA protected the primary follicles in the ovary from γ-radiation to the same extent as loss of p63.86 This demonstrates that p63 induces apoptosis in the same way as p53. Remarkably, PUMA/NOXA double knockout and even PUMA single knockout females, when irradiated either as pups or as adults, retained normal fertility. Of note, among several hundred offspring of such irradiated Puma-/-Noxa-/- or Puma-/- mothers, none were found to exhibit developmental abnormalities or cancer predisposition.86 This means that primordial follicles that are protected from γ-radiation-induced apoptosis due to the absence of PUMA or PUMA plus NOXA must be able to repair their DNA lesions highly efficiently.
Of note, the nematode C. elegans homologue of p53/p63/p73 also has a function in DNA damage-induced killing of female germ cells and it induces a pathway to apoptosis that is initiated by the pro-apoptotic BH3-only protein, EGL-1, and can be inhibited by the pro-survival BCL-2 homologue, CED-9.87 This demonstrates that DNA damage-induced, p63-induced apoptosis via the BCL-2-regulated pathway in female germ cells is evolutionarily highly conserved and is likely to play a critical role in safeguarding genomic stability in the germline.88
Impact of p53 on the Death Receptor Apoptotic Pathway
Importantly, p53 can also regulate the expression of components of the extrinsic apoptotic pathway. The p53 can transcriptionally induce the genes encoding FAS (also called APO-1 and CD95) and possibly other genes encoding related death receptors.89 Some studies have reported that cytotoxic drugs that cause activation of p53 (e.g., etoposide, cyclophosphamide) and γ-radiation can induce apoptosis in leukaemic cells through the death receptor pathway.90 However, experiments using a panel of transgenic and gene knockout mice demonstrated beyond doubt that DNA damage-inducing anticancer drugs and γ-radiation kill normal as well as transformed cells by activating the BCL-2-regulated apoptotic pathway91, 92 in a p53-dependent manner. In striking contrast, complete loss of the death receptor apoptotic pathway (e.g., due to loss of function of caspase-8 or its activator FADD) does not protect these cells from these agents.44, 93 Although we would conclude that p53-mediated upregulation of death receptors is not essential for cell killing, it may serve to sensitise cells to so-called death ligands (e.g., FASL, TNF; the ligands for the death receptors FAS and TNFR1) expressed on neighbouring cells. This would allow for paracrine killing by cytotoxic T cells or NK cells and such a process may contribute to the effectiveness of cancer therapy in certain cancers.
Indirect Effects of p53 on Apoptosis Signalling
To further add to the complexity of p53-mediated control of apoptosis, p53 also drives the expression of several genes whose functions do not lie within the two apoptotic pathways per se but may modulate the cellular response to cell death-inducing insults. For example, p53 drives expression of a number of microRNA species, including miR-34,94 that is known to target the pro-survival Bcl-2 gene.95 Thus, p53-induced miR34a expression may sensitise cells to apoptotic stimuli by reducing the levels of BCL-2. Another well-characterised transcriptional target of TP53 is Zmat396 that has a poorly defined function but has been shown to impact on the response of cells to apoptotic stimuli.97 Of note, p53 can also drive the expression of various genes that may serve pro-survival functions, such as BTG2 and PLK2.96 Thus, p53 signalling should not be viewed as exclusively inducing apoptosis, but in certain situations p53 activation may preferentially activate processes that enhance cell survival and cell growth. This may well be pertinent to tumour development and cancer therapy.
Reported Roles of p53 in Other Forms of Cell Death
Although the role of p53 (and p63) in the induction of apoptosis is widely accepted, there are also reports that p53 can regulate additional non-apoptotic cell death pathways. For example, p53 was reported to open the mitochondrial permeability transition pore to thereby induce necrotic cell death.98 Moreover, p53 has also been reported to sensitise cells to ferroptosis, a non-apoptotic form of cell death,99 by repressing expression of SLC7A11, a key component of the cystine/glutamate antiporter.100 However, the relevance of these processes to normal physiology (e.g., the death of nontransformed cells with DNA lesions) or cancer therapy-induced killing of tumour cells has not been established.
Future Directions in Research on p53-Induced Cell Death
A ‘holy grail’ in research on p53 is to understand the mechanistic basis determining the strongly context-dependent functional output following p53 activation. For example, p53 activation by nutlin-3a results in apoptosis in some cells but cell cycle arrest and senescence in others (both malignant and nontransformed).67 Moreover, restoration of wild-type p53 in cancers driven by loss of p53 (plus additional oncogenic lesions) causes apoptosis in lymphoma cells but cell senescence and cell cycle arrest in solid organ cancers.101, 102, 103, 104 Importantly, there is evidence that p53 activation and expression may occur without necessarily resulting in apoptosis or senescence, as has been observed in stem cells, where p53 activation may drive differentiation rather than exerting antiproliferative effects.105 Finally, dramatic differences are seen in the sensitivity of different cell types to p53 activation. For example, cells within the gastrointestinal tract106 and the haematopoietic system54, 55, 91 are particularly vulnerable to p53-induced apoptosis that underlies their prominent involvement in toxicity associated with DNA damage-inducing chemotherapy. We can speculate on factors that may differentiate the p53 response: post-translational modifications on the p53 protein (e.g., acetylation, phosphorylation) that alter the function of p53 as a transcription factor, functional interaction between p53 and other transcription factors that may vary according to cell type, context-dependent regulation of specific target genes (e.g., altered regulation in the setting of oncogene expression), epigenetic regulation of p53 target genes (e.g., p53 regulation may vary depending on whether certain target genes are epigenetically silenced) as well as post-transcriptional and post-translational regulation of p53 target genes or their protein products (e.g., a role for p53-independent microRNA-mediated regulation). These are all fascinating possibilities that require much further investigation.
The Role of p53-Induced Apoptosis in p53-Mediated Tumour Suppression
The finding that p53 can induce apoptosis led to the widely accepted assumption that out of all the cellular effector processes activated by p53 (Figure 3) this is the most critical, possibly even the sole, process by which p53 suppresses tumour development (reviewed in Vousden and Lane9). This made sense: after all, if cells during the early stages of neoplastic transformation are killed through p53-induced apoptosis, no fully transformed malignant cells will emerge from this clone. However, matters are not that simple. If apoptosis is the critical process for p53-mediated tumour suppression, it would be predicted that Puma-/-Noxa-/- mice should be as prone to spontaneous or oncogene-driven tumour development as Trp53-/- mice themselves, as combined loss of PUMA and NOXA abrogates p53-induced apoptosis in many (possibly all) cell types.64 Contrary to this prediction, no spontaneous tumour development was observed in a large cohort of Puma-/-Noxa-/- mice that were monitored over a long time period.64, 107 Induction of G1/S boundary cell cycle arrest and cell senescence are also thought to be processes that could be critical for p53 tumour suppression (Figure 3). The CDK inhibitor p21 is essential for cell cycle arrest and also a major contributor to cellular senescence.108 It is therefore remarkable that mice with mutations in Trp53 that impair its ability to transcriptionally induce Puma, Noxa and p21 and even mice completely deficient for these genes (i.e., Puma-/-Noxa-/-p21-/- mice) do not spontaneously develop any tumours (Figure 4).107, 109, 110 Notably, the cells from all of these mutant mice are unable to undergo apoptosis, cell cycle arrest or senescence upon p53 activation. This demonstrates that p53 is capable of preventing spontaneous development of cancer in the complete absence of its ability to induce apoptosis, G1/S cell cycle arrest and cell senescence. Moreover, combined loss of PUMA and p21 or mutations in the two transactivation domains of p53 that are critical for the transcriptional induction of Puma, Noxa and p21 accelerate c-MYC-driven lymphoma development and mutant RAS-driven lung cancer development to a much lesser extent than loss of p53 (Figure 4).111, 112 The observation that loss of PUMA (or combined loss of PUMA and NOXA or PUMA and p21) can accelerate c-MYC-driven lymphoma development112, 113 does, however, show that p53-mediated apoptosis (via PUMA and NOXA) can exert significant tumour suppressive function. The relative importance of the induction of apoptosis to overall TP53-mediated tumour suppression is likely to vary depending on the type of cell undergoing neoplastic transformation and the nature of the oncogenic lesions that drive tumorigenesis.

P53 activates a multitude of cellular effector processes. Model showing a selected cellular effector processes that can be activated by p53. Some, but not all, of the p53 target genes that are critical for the execution of these processes are indicated. The challenge remains to understand of which of these processes are critical for tumour suppression in which setting; that is, cell of origin undergoing neoplastic transformation and nature of the oncogenic lesions driving their transformation

Impact of p53-induced apoptosis on tumour development. Models showing the impact of loss of p53-induced apoptosis on tumour development in different cancer models/settings
Important insight into the mechanisms that are critical for TP53-mediated tumour suppression also came from experiments using an elegant genetically engineered mouse model in which p53 activity can be turned on or off at will.114 These studies used the γ-radiation-induced thymic T-cell lymphoma model and showed that the presence of p53 during the acute response to DNA damage (characterised by extensive apoptosis of many haematopoietic cell types) was not needed for tumour suppression. Instead, p53 function was required during the later recovery phase. Accordingly, p53-mediated tumour suppression was not activated through the DNA damage sensing process but instead through p19/ARF114 that is activated in response to the expression of oncogenes (e.g., deregulated c-MYC expression). P19/ARF inhibits MDM2, the major negative regulator of p53. This indicates that in this model, thymic T-cell lymphomas emerge from the stem/progenitor cells that have sustained potentially oncogenic DNA lesions and are mobilised to replenish the haematopoietic system that was depleted by γ-radiation. The very rapid proliferation of progenitor cells bearing oncogenic lesions, which is likely to also be the basis of the development of many other cancers, may facilitate the acquisition of mutations in oncogenes or suppressor genes that further drive neoplastic transformation. Some initially paradoxical findings are consistent with this. In the aforementioned γ-radiation-induced thymic T-cell lymphoma development mouse model, loss of pro-apoptotic PUMA or overexpression of pro-survival BCL-2 completely prevented tumour development (Figure 4).115, 116 The explanation for this is that loss of pro-apoptotic PUMA or pro-survival BCL-2 overexpression prevented the γ-radiation-induced death of diverse leukocyte populations and this obviated the need for mobilisation and excess proliferation of haematopoietic stem/progenitor cells in the bone marrow that are thought to be the lymphoma/leukaemia-initiating cancer stem cells in this model.117 This concept may extend to many additional cancers, including epithelial ones, as overexpression of pro-survival BCL-2 paradoxically delays liver cancer development,118 whereas, conversely, loss of pro-survival MCL-1 promotes its development.119
Impact of Mutant p53 on WT p53-Induced Apoptosis and on WT p53-Mediated Tumour Suppression
The p53 is unusual among tumour suppressors. In tumours that are driven by mutations in the tumour suppressors PTEN or RB, the expression of these proteins is usually lost completely because of the nature of the mutations selected for during tumorigenesis (reviewed in Knudsen and Knudsen120 and Yin and Shen121). In contrast, many tumours that are driven by mutations in TP53 express high levels of the mutant p53 protein and show a loss of the other allele of TP53 (reviewed in Vousden and Lane9 and Freed-Pastor and Prives10). In fact, the high-level mutant p53 protein expression can be used as a diagnostic marker for cancers driven by mutations in the TP53 gene (reviewed in Liu and Gelmann122). The highly expressed mutant p53 protein can promote tumorigenesis in three ways: (1) loss of the WT p53 activity, (2) DNEs over the WT p53 protein early in transformation before loss of the WT TP53 allele, through the formation of mixed tetramers containing both wild-type and mutant p53 proteins and (3) de novo GOFs that are mediated through interactions of mutant p53 protein with other transcription factors and tumour suppressors (e.g., p63, p73) (Figure 5) (reviewed in Vousden and Lane9 and Freed-Pastor and Prives10). As early in transformation, mutant p53 levels are often variable and low,123 it appears likely that the GOF effects may only come into play at a late stage of transformation. It is obvious how loss of the WT p53 function contributes to the tumour promoting action of mutant p53 but the mechanisms by which the DNE and GOF effects of mutant p53 drive tumour development are not established. A detailed understanding of the role of these processes in the development as well as the sustained growth of tumours is anticipated to identify targetable vulnerabilities for the development of novel cancer therapies.

Impact of mutant p53 proteins on tumour development. Model depicting the mechanisms by which mutant p53 proteins, which are frequently highly overexpressed (compared with the wild-type p53 protein), on tumour development
Conclusions
In conclusion, TP53 is arguably one of the most important (if not the most important) genes in human cancer. It appears that p53 is critical for tumour suppression not during the acute response to cellular stress, such as DNA damage (e.g. caused by γ-radiation and reliant on the CHK1/CHK2 kinases) that is characterised by extensive apoptosis, but for the killing or silencing of the cancer-initiating (often thought to be stem/progenitor) cells that have acquired oncogenic lesions that drive the neoplastic transformation. The p53 transcription factor activates several effector processes, apoptotic cell death being one of them. Contrary to long-held perceptions, loss of p53-induced apoptosis (via PUMA and NOXA) even when combined with additional loss of induction of G1/S boundary cell cycle arrest and cell senescence (via p21) does not lead to spontaneous tumour development, in striking contrast to loss or mutation of p53. Thus, additional cellular processes, either by themselves or in a manner overlapping with the aforementioned mechanisms, must account for the potent tumour suppressive action of p53. Identifying these signalling pathways and how they are integrated will provide exciting research opportunities for several years. A better understanding of p53-mediated apoptosis and p53-mediated tumour suppression more generally holds promise for various potential clinical applications. These include improving the efficacy of anticancer therapies that rely on p53 activation, reducing the toxicities associated with chemotherapy and radiotherapy and improving haematopoietic stem cell transplant conditioning regimens and perhaps also in nonmalignant settings where abnormal induction of cell death pathways (that may in part be driven by p53) contributes to tissue damage, such as myocardial infarction and cerebral ischaemia.
References
Levine AJ, Momand J, Finlay CA . The p53 tumour suppressor gene. Nature 1991; 351: 453–456.
Lane DP, Benchimol S . p53: oncogene or antioncogene. Genes Dev 1990; 4: 1–8.
Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M . Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci USA 1989; 86: 8763–8767.
Finlay CA, Hinds PW, Levine AJ . The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989; 57: 1083–1093.
Hollstein M, Sidransky D, Vogelstein B, Harris CC . p53 mutations in human cancers. Science 1991; 253: 49–53.
Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B . Suppression of human colorectal carcinoma cell growth by wild-type p53. Science 1990; 249: 912–915.
Malkin D, Li FP, Strong LC, Fraumeni JFJ, Nelson CE, Kim DH et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250: 1233–1238.
Srivastava S, Zou ZQ, Pirollo K, Plattner W, Chang EH . Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 1990; 348: 747–749.
Vousden KH, Lane DP . p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8: 275–283.
Freed-Pastor WA, Prives C . Mutant p53: one name, many proteins. Genes Dev 2012; 26: 1268–1286.
Reich NC, Levine AJ . Growth regulation of a cellular tumour antigen, p53, in nontransformed cells. Nature 1984; 308: 199–201.
Haupt Y, Maya R, Kazaz A, Oren M . Mdm2 promotes the rapid degradation of p53. Nature 1997; 387: 296–299.
Kubbutat MH, Jones SN, Vousden KH . Regulation of p53 stability by Mdm2. Nature 1997; 387: 299–303.
Shieh S-Y, Ikeda M, Taya Y, Prives CDN . DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91: 325–334.
Oren M . Regulation of the p53 tumor suppressor protein. J Biol Chem 1999; 274: 36031–36034.
Riley T, Sontag E, Chen P, Levine A . Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9: 402–412.
Kamada R, Toguchi Y, Nomura T, Imagawa T, Sakaguchi K . Tetramer formation of tumor suppressor protein p53: structure, function, and applications. Biopolymers 2016; 106: 598–612.
Laptenko O, Prives C . Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ 2006; 13: 951–961.
Menendez D, Inga A, Resnick MA . The expanding universe of p53 targets. Nat Rev Cancer 2009; 9: 724–737.
Cho Y, Gorina S, Jeffrey PD, Pavletich NP . Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994; 265: 346–355.
Abbas T, Dutta A . p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009; 9: 400–414.
Sengupta S, Harris CC . p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 2005; 6: 44–55.
Michael D, Oren M . The p53 and Mdm2 families in cancer. Curr Opin Genetics Dev 2002; 12: 53–59.
Montes de Oca Luna R, Wagner DS, Lozano G . Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378: 203–206.
Jones SN, Roe AE, Donehower LA, Bradley A . Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378: 206–208.
Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M . Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991; 352: 345–347.
Johnson P, Chung S, Benchimol S . Growth suppression of Friend virus-transformed erythroleukemia cells by p53 protein is accompanied by hemoglobin production and is sensitive to erythropoietin. Mol Cell Biol 1993; 13: 1456–1462.
Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J . Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci USA 1992; 89: 4495–4499.
Ramqvist T, Magnusson KP, Wang Y, Szekely L, Klein G, Wiman KG . Wild-type p53 induces apoptosis in a Burkitt lymphoma (BL) line that carries mutant p53. Oncogene 1993; 8: 1495–1500.
Strasser A, O'Connor L, Dixit VM . Apoptosis signaling. Annu Rev Biochem 2000; 69: 217–245.
Strasser A, Harris AW, Huang DCS, Krammer PH, Cory S . Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J 1995; 14: 6136–6147.
Green DR . Apoptotic pathways: ten minutes to dead. Cell 2005; 121: 671–674.
Czabotar PE, Lessene G, Strasser A, Adams JM . Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014; 15: 49–63.
Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N et al. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ 2012; 19: 915–925.
Ke F, Bouillet P, Kaufmann T, Strasser A, Kerr J, Voss AK . Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis 2013; 4: e650.
Ke F, Grabow S, Kelly GL, Lin A, O'Reilly LA, Strasser A . Impact of the combined loss of BOK, BAX and BAK on the hematopoietic system is slightly more severe than compound loss of BAX and BAK. Cell Death Dis 2015; 6: e1938.
Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S et al. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 2016; 165: 421–433.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES et al. Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479–489.
Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998; 94: 325–337.
Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 1998; 94: 339–352.
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P . Apaf-1 (CED-4 homologue) regulates programmed cell death in mammalian development. Cell 1998; 94: 727–737.
Yoshida H, Kong Y-Y, Yoshida R, Elia AJ, Hakem A, Hakem R et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998; 94: 739–750.
Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998; 9: 267–276.
Strasser A, Jost PJ, Nagata S . The many roles of FAS receptor signaling in the immune system. Immunity 2009; 30: 180–192.
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998; 17: 1675–1687.
Li H, Zhu H, Xu C-J, Yuan J . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501.
Luo X, Budlhardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl-2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
Yin X-M, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999; 400: 886–891.
Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009; 460: 1035–1039.
Wang Y, Szekely L, Okan I, Klein G, Wiman KG . Wild-type p53-triggered apoptosis is inhibited by bcl-2 in a v-myc-induced T-cell lymphoma line. Oncogene 1993; 8: 3427–3431.
Strasser A, Harris AW, Jacks T, Cory S . DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell 1994; 79: 329–339.
Chiou S-K, Rao L, White E . Bcl-2 blocks p53-dependent apoptosis. Mol Cell Biol 1994; 14: 2556–2563.
Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T . p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993; 362: 847–849.
Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML et al. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 1993; 362: 849–852.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T et al. Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000; 288: 1053–1058.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B . PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7: 673–682.
Nakano K, Vousden KH . PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7: 683–694.
Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ et al. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci USA 2001; 98: 11318–11323.
Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A et al. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev 2003; 17: 2233–2238.
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003; 302: 1036–1038.
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003; 4: 321–328.
Naik E, Michalak EM, Villunger A, Adams JM, Strasser A . UV-radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J Cell Biol 2007; 176: 415–424.
Michalak EM, Villunger A, Adams JM, Strasser A . In several cell types the tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ 2008; 15: 1019–1029.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17: 393–403.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 2005; 17: 525–535.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848.
Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ et al. Maximal killing of lymphoma cells by DNA-damage inducing therapy requires not only the p53 targets Puma and Noxa but also Bim. Blood 2010; 116: 5256–5267.
Valente LJ, Aubrey BJ, Herold MJ, Kelly GL, Happo L, Scott CL et al. Therapeutic response to non-genotoxic activation of p53 by Nutlin3a is driven by PUMA-mediated apoptosis in lymphoma cells. Cell Rep 2016; 14: 1858–1866.
O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J 1998; 17: 384–395.
Bouillet P, Metcalf D, Huang DCS, Tarlinton DM, Kay TWH, Köntgen F et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 1999; 286: 1735–1738.
Li M, He Y, Dubois W, Wu X, Shi J, Huang J . Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell 2012; 46: 30–42.
Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 2013; 27: 1016–1031.
Tonelli C, Morelli MJ, Bianchi S, Rotta L, Capra T, Sabo A et al. Genome-wide analysis of p53 transcriptional programs in B cells upon exposure to genotoxic stress in vivo. Oncotarget 2015; 6: 24611–24626.
Selvakumaran M, Lin H-K, Miyashita T, Wang HG, Krajewski S, Reed JC et al. Immediate early up-regulation of bax expression by p53 but not TGFb1: a paradigm for distinct apoptotic pathways. Oncogene 1994; 9: 1791–1798.
Thornborrow EC, Patel S, Mastropietro AE, Schwartzfarb EM, Manfredi JJ . A conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax genes. Oncogene 2002; 21: 990–999.
Robles AI, Bemmels NA, Foraker AB, Harris CC . APAF-1 is a transcriptional target of p53 in DNA damage-induced apoptosis. Cancer Res 2001; 61: 6660–6664.
Rathmell JC, Lindsten T, Zong W-X, Cinalli RM, Thompson CB . Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol 2002; 3: 932–939.
Marsden V, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert P et al. Apoptosis initiated by Bcl–2-regulated caspase activation independently of the cytochrome c/Apaf–1/caspase–9 apoptosome. Nature 2002; 419: 634–637.
Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK et al. Developmental regulation of mitochondrial apoptosis by c-Myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell 2017; 31: 142–156.
Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2: 305–316.
Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997; 90: 809–819.
Yang A, Kaghad M, Caput D, McKeon F . On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet 2002; 18: 90–95.
Jost CA, Marin MC, Kaelin WG Jr. . p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 1997; 389: 191–194.
Suh EK, Yang A, Kettenbach A, Bamberger C, Michaelis AH, Zhu Z et al. p63 protects the female germ line during meiotic arrest. Nature 2006; 444: 624–628.
Kerr JB, Hutt KJ, Michalak EM, Cook M, Vandenberg CJ, Liew SH et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol Cell 2012; 48: 343–352.
Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L et al. Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr Biol 2002; 12: 1908–1918.
Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G . The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol 2011; 12: 259–265.
Müller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 1998; 188: 2033–2045.
Friesen C, Herr I, Krammer PH, Debatin K-M . Involvement of the CD95 (APO-1/Fas) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat Med 1996; 2: 574–577.
Strasser A, Harris AW, Cory S . Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 1991; 67: 889–899.
Miyashita T, Reed JC . bcl-2 gene transfer increases relative resistance of S49.1 and WEHI7.2 lymphoid cells to cell death and DNA fragmentation induced by glucocorticoids and multiple chemotherapeutic drugs. Cancer Res 1992; 52: 5407–5411.
Newton K, Strasser A . Ionizing radiation and chemotherapeutic drugs induce apoptosis in lymphocytes in the absence of fas or FADD/MORT1 signaling: implications for cancer therapy. J Exp Med 2000; 191: 195–200.
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447: 1130–1134.
Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 2007; 17: 1298–1307.
Allen MA, Andrysik Z, Dengler VL, Mellert HS, Guarnieri A, Freeman JA et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife 2014; 3: e02200.
Bersani C, Xu LD, Vilborg A, Lui WO, Wiman KG . Wig-1 regulates cell cycle arrest and cell death through the p53 targets FAS and 14-3-3sigma. Oncogene 2014; 33: 4407–4417.
Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM . p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012; 149: 1536–1548.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149: 1060–1072.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015; 520: 57–62.
Martins CP, Brown-Swigart L, Evan GI . Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006; 127: 1323–1334.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445: 656–660.
Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445: 661–665.
Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010; 468: 567–571.
Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 2005; 7: 165–171.
Merritt AJ, Potten CS, Kemp CJ, Hickman JA, Balmain A, Lane DP et al. The role of p53 in spontaneous and radiation-induced apoptosis in the gastrointestinal tract of normal and p53-deficient mice. Cancer Res 1994; 54: 614–617.
Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL et al. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 2013; 3: 1339–1345.
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P . Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82: 675–684.
Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011; 145: 571–583.
Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012; 149: 1269–1283.
Jiang D, Brady CA, Johnson TM, Lee EY, Park EJ, Scott MP et al. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc Natl Acad Sci USA 2011; 108: 17123–17128.
Valente LJ, Grabow S, Vandenberg CJ, Strasser A, Janic A . Combined loss of PUMA and p21 accelerates c-MYC-driven lymphoma development considerably less than loss of one allele of p53. Oncogene 2016; 35: 3866–3871.
Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ 2009; 16: 684–696.
Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI . The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 2006; 443: 214–217.
Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J et al. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev 2010; 24: 1602–1607.
Michalak EM, Vandenberg CJ, Delbridge ARD, Wu L, Scott CL, Adams JM et al. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev 2010; 24: 1608–1613.
Kaplan HS, Brown MB . Protection against radiation-induced lymphoma development by shielding and partial-body irradiation of mice. Cancer Res 1952; 12: 441–444.
Pierce RH, Vail ME, Ralph L, Campbell JS, Fausto N . Bcl-2 expression inhibits liver carcinogenesis and delays the development of proliferating foci. Am J Pathol 2002; 160: 1555–1560.
Weber A, Boger R, Vick B, Urbanik T, Haybaeck J, Zoller S et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 2010; 51: 1226–1236.
Knudsen ES, Knudsen KE . Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 2008; 8: 714–724.
Yin Y, Shen WH . PTEN: a new guardian of the genome. Oncogene 2008; 27: 5443–5453.
Liu MC, Gelmann EP . P53 gene mutations: case study of a clinical marker for solid tumors. Semin Oncol 2002; 29: 246–257.
Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004; 119: 861–872.
Acknowledgements
We thank all of our current and past colleagues at The Walter and Eliza Hall Institute and elsewhere. Our work is supported by the Australian National Health and Medical Research Council (Program Grant 1016701 to AS and Fellowship 1020136 1020363 to AS), the Leukemia and Lymphoma Society of America (LLS SCOR 7001-13 to AS), the Cancer Council of Victoria (1052309 to AS) as well as operational infrastructure grants through the Australian Government Independent Research Institute Infrastructure Support Scheme (9000220) and the Victorian State Government Operational Infrastructure Support Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by F Pentimalli
Rights and permissions
About this article

Cite this article
Aubrey, B., Kelly, G., Janic, A. et al. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression?. Cell Death Differ 25, 104–113 (2018). https://doi.org/10.1038/cdd.2017.169
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.169
This article is cited by
-
Network pharmacology study to explore the multiple molecular mechanism of SH003 in the treatment of non-small cell lung cancer
BMC Complementary Medicine and Therapies (2024)
-
Simultaneous inhibition of FAK and ROS1 synergistically repressed triple-negative breast cancer by upregulating p53 signalling
Biomarker Research (2024)
-
Bifidobacterium longum SX-1326 ameliorates gastrointestinal toxicity after irinotecan chemotherapy via modulating the P53 signaling pathway and brain-gut axis
BMC Microbiology (2024)
-
Combined absence of TRP53 target genes ZMAT3, PUMA and p21 cause a high incidence of cancer in mice
Cell Death & Differentiation (2024)
-
Genome-wide CRISPR screens identify PKMYT1 as a therapeutic target in pancreatic ductal adenocarcinoma
EMBO Molecular Medicine (2024)
