
Abstract
The receptor for epidermal growth factor (EGFR) is a prime target for cancer therapy across a broad variety of tumor types. As it is a tyrosine kinase, small molecule tyrosine kinase inhibitors (TKIs) targeting signal transduction, as well as monoclonal antibodies against the EGFR, have been investigated as anti-tumor agents. However, despite the long-known enigmatic EGFR gene amplification and protein overexpression in glioblastoma, the most aggressive intrinsic human brain tumor, the potential of EGFR as a target for this tumor type has been unfulfilled. This review analyses the attempts to use TKIs and monoclonal antibodies against glioblastoma, with special consideration given to immunological approaches, the use of EGFR as a docking molecule for conjugates with toxins, T-cells, oncolytic viruses, exosomes and nanoparticles. Drug delivery issues associated with therapies for intracerebral diseases, with specific emphasis on convection enhanced delivery, are also discussed.
Similar content being viewed by others


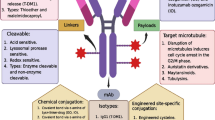
Targeting the EGFR signal transduction pathway faces the issue of redundant alternative signaling pathway activation and rapid adaptation. |
EGFR expression is highly variable within a glioblastoma. |
Intracompartmental cell surface targeting with large effector molecules or viral agents holds most promise to overcome the therapeutic deadlock. |
1 Introduction
Targeting the receptor for epidermal growth factor (EGFR) has been rewarding in cancer and many pharmaceuticals are approved alone or in combination with chemotherapy for colorectal cancer, non-small-cell lung cancer, and pancreatic cancer, among others, but not for gliomas [1]. The approved agents are mostly tyrosine kinase inhibitors (TKIs) interfering with the receptor signaling, or monoclonal antibodies targeting the receptor at the cell surface to interfere with ligand binding (Fig. 1). It remains unresolved why EGFR targeting has not been successful for glioma as it should be ideally suitable in the context of this disease. EGFR was the first molecule to be linked to oncogenesis in glioblastoma [2]. During the time of the first oncogene descriptions, its gene became linked to a viral oncogene—v-erb B. Massive amplification of that gene was found in glioblastoma [3] and somatic copy number alterations are present in 43% of patients [4]. In addition, subsequently numerous mutations including constitutively active truncations and an in-frame deletion leading to constitutive activation of the intracellular tyrosine kinase were described as well as the numerous diverse intracellular signaling consequences [5]. Overall, about 60% of glioblastoma patients have some kind of genomic alteration affecting this pathway [4]. Of particular interest became the vIII mutation, which results in a molecule with an altered amino-acid sequence, giving rise to a unique site of antigenicity [6]. In many correlative analyses of EGFR status in clinical trials for glioblastoma, it was reported to be prognostically relevant [7], although a larger meta-analysis failed to confirm that overall [8]. In all large genome-wide cancer studies it turned out to be a key molecule for glioma [9] as well as for other tumor entities. Therefore, because of being a ‘signature molecule’ for glioblastoma, EGFR was thought to be an ideal target for therapy [10, 11].

Integrative sketch of epidermal growth factor receptor (EGFR) targeted treatment modalities and additional technologies. Focused ultrasound may be combined with EGFR-targeted nanoparticles to result in local release of cargo; likewise, boronated EGFR binding compounds will only be active (small flashes) in the field of a neutron beam. The sketch also illustrates the heterogeneity of the different types of EGFR expression including the mutation types and amplification patterns. The tumor is made up of cells heterogeneous in their EGFR expression and alterations as indicated by the different cell types (see text). To improve unsatisfactory intravenous delivery, delivery of large molecules or even viruses to the tumor (dark pink) or the invasive zone (light pink) convection (CED) is a suitable technique as indicated by the two porous catheter tips in the top part of the figure. BBB blood–brain barrier, BNCT boron neutron capture therapy, CAR chimeric antigen receptor, EGFRvIII epidermal growth factor receptor variant III, EGFRwt/mut epidermal growth factor receptor wild-type/mutant, mABs monoclonal antibodies, RTK receptor tyrosine kinase, HSR homogeneously staining region
There are many possible explanations besides drug delivery issues for the still disappointing exploration of EGFR as a target for brain tumors, including a multitude of adaptive mechanisms [12], alternate pathways adaptation, and loss of relevance in later disease stages. These come to bear mostly with agents interfering with receptor signaling in the attempt to interrupt the activation of proliferative or migratory programs. These are mostly small molecules, so-called TKIs or monoclonal antibodies.
Alternatively, the EGFR has also been exploited as a target to deliver therapeutics to the tumor which are intrinsically toxic and thus independent from the activated signaling pathway, or trigger other processes like immune activation. Such constructs can be targeted toxins with an EGFR-binding ligand linked to a toxic molecule, which then relies on receptor internalization for specific delivery, or chimeric antigen receptor T cells which also recognize the EGFR as a docking molecule. The paradigmatic approaches are briefly summarized in Table 1.
Whatever the nature of the EGFR targeting agent, all face the problem of delivery, so aside from the adaptive mechanisms mentioned, delivery problems across the blood–brain barrier is another often-cited explanation for failure of EGFR targeting for glioblastoma.
That leads to the issue of local intraparenchymal or, ideally, compartmental delivery.
2 Compartmental Selectivity of EGFR Targeting
Direct compartmental targeting of EGFR in the brain in the context of intrinsic brain tumors is attractive from a drug safety standpoint because of its high degree of selectivity for the disease in the brain. In normal brain cells, be they neurons, normal glial cells, or microglia, EGFR is of little relevance. It was found on endothelial cells of the brain in elderly patients [13] and is thought to play a role earlier in development with a predominantly neural expression, mostly in the cerebellum. The molecule could also be detected in the adult hippocampus [14], so there may also be expression on neuro-glial stem cells. Most of the neurobiological context of the EGF/EGFR system has been worked out in rodents [15] and that should inform many of the reservations with strategies against EGFR in the brain in humans, which are almost exclusively pursued in the context of intrinsic brain tumors. In the rodent brain, for example, presence of a functioning EGFR is thought to be necessary for the interaction of neural stem cells with their environment to maintain their self-renewal capabilities [16]. There is little direct evidence in humans for this [17], but in a rare investigation of human tissues over a wide age range, it appeared as if EGF and EGFR are relevant for a microglial sustained persistence or proliferation of subventricular zone neuroglial stem cells [18]. Circumstantial support may come from the limited experience using EGFR TKIs in addition to radiation for brain metastases, which resulted in increased survival but was associated with increased neurocognitive decline [19], possibly an effect on stem-cell-based regenerative capacities. In contrast, a more localized approach with EGFR inhibition using HER-2/EGFR TKIs concurrent to stereotactic radiosurgery for brain metastasis significantly decreased 12-month incidence of local failure [20] without such sequelae.
Apart from vascular endothelial cells and neuroglial stem cells, EGFR is also expressed on meningeal cells and consequently on meningiomas [21]. The expression of EGFR has nevertheless not been found to be related to the clinical course [22] and no therapy has emerged from that finding [23].
Taken together, the (EGF-TGFα [tumor growth factor-alpha])/EGFR system appears to have a rather limited role in the adult central nervous system (CNS) so that a high degree of selectivity of the EGFR expression to glial neoplasia within the brain exists. Therefore, compartmental drug delivery arises as a valid, major strategy to increase efficacy of EGFR-based therapies.
3 EGFR Targeting Agents
3.1 Small Molecule Kinase Inhibitors
Among the many agents developed to target the EGFR, the so-called small molecule TKIs interfere with the signal transduction cascade of its tyrosine kinase activity [24]. Despite the availability of several compounds that are approved for a broad spectrum of diseases, none is approved for glioblastoma, which is a result of numerous negative clinical trials. For the leading representatives of this group, erlotinib, gefitinib, afatinib, and lapatinib, trials have not shown efficacy either alone or in combination. Erlotinib showed no efficacy and unacceptable side effects as a single agent in newly diagnosed glioblastoma [25] and later studies combining it for recurrent glioblastoma with mechanistic target of rapamycin (mTOR) blockers or bevacizumab were unsuccessful [26,27,28]. Gefitinib did not result in improved overall survival in a phase II trial in recurrent glioblastoma [29] or in a phase I/II trial in combination with radiation in newly diagnosed glioblastoma [30]. Afatinib as a single agent has shown only limited efficacy in one trial in patients with recurrent glioblastoma [31]. Lapatinib has shown very limited efficacy as a single agent in an early clinical trial in combination with pazopanib, an oral anti-angiogenic TKI [32], which as a single agent also showed no efficacy in recurrent glioblastoma [33].
One of the issues is brain penetrance, which has been a specific aim for the chemical design of new reagents for which there is only preclinical promise [34]. Brain penetrance is difficult to measure reliably and only recently complex mass spectrometric methods have become available that, at least in animal models, promise to give precise estimates of substance distribution and metabolization. At least for erlotinib it could be shown that it distributes inside an intracranial U87 xenograft [35]. Earlier correlative pharmacokinetic studies of two clinical trials with erlotinib and gefitinib, from which tissue was available from recurrence/progression surgery, showed that erlotinib only accumulated 5–7% of the plasma concentration in the tumor. Gefitinib, in contrast, was sequestered in the tissue and reached 2- to 3-fold plasma concentrations so that, at least for gefitinib, lack of concentration could not have been the cause of insufficient efficacy [36].
Another issue is the lack of specificity, as the TKIs often act on several tyrosine kinases, albeit with differential efficacy. The adaptive capacity of glioblastoma cells, which activate a multiplicity of redundant pathways, is rapid and therefore easily overcomes inhibition of just one of them [37, 38]. This causes toxicity in the absence of efficacy as the normal cells lack that redundancy of pathway activation.
3.2 Monoclonal Antibodies
The drug development of monoclonal antibodies recognizing EGFR for the treatment of glioblastoma is more complex as they can serve not only as agents interfering with ligand binding, thus inactivating signaling, but can be used in the context of targeting drugs via conjugates and internalization or work by immunological mechanisms. Under normal circumstances, a cognate ligand binds to EGFR and that association leads to recruitment of that complex to clathrin-coated pits with subsequent internalization and downregulation of the receptor from the cell surface [39, 40]. In that context, antibodies to EGFR show a broad spectrum of effects. They can be either blocking antibodies which prevent ligand binding and can be receptor activating or non-activating, and induce internalization or not. Antibodies which are blocking and internalized are ideal for downregulating the receptor. They are also suitable to ferry conjugates like toxins into the cells. Cell surface binding antibodies which are not internalized will either disrupt ligand-mediated signaling or lead to activation of immunologic mechanisms like antibody-dependent cell-mediated cytotoxicity (ADCC).
Several antibodies developed against EGFR have been used against glioblastoma. Cetuximab was first approved for colorectal cancer and is an antibody-blocking ligand binding without receptor activation [41]. In a preclinical study with glioblastoma xenografts, the intraparenchymal delivery of cetuximab together with systemic application of an anti-angiogenic antibody against the receptor for vascular endothelial growth factor (VEGF) resulted in a marked reduction of tumor cell migration/invasion seen after VEGF inhibition [42] The relevance of EGFR for glioma cell migration has long been known [43] and accordingly, the highest expression of EGFR was demonstrated on the infiltrative edge of tumors [44, 45]. Whether this indicates a relationship between the migratory program of neural stem cells remains to be proven [46]. The direct connection between EGFR and migration of glioma cells offers a target opportunity. It was seen that CSN6, part of a regulator of the ubiquitin-proteasome-dependent degradation of cancer-related proteins such as p53, c-myc, and c-Jun, when overexpressed reduces the degradation of EGFR and thus enhances the migratory and invasive properties of glioblastoma [47].
In clinical trials, cetuximab failed to demonstrate efficacy either as a single agent for recurrent glioblastoma [48] or in combination with other reagents [49] or radiation [50] and therefore was clinically not developed beyond phase II. Nonetheless, the agent holds promise to be developed further and circumventing the delivery issue of large molecules by direct intracranial infusion, cetuximab effectively blocked glioblastoma cell invasion in an orthotopic xenograft model [42]. Another intracranial strategy being developed is also in preclinical stages where an adenoviral delivery of the corresponding antibody gene by direct intracerebral injection was used so that the antibody would be produced by the transduced cells to increase the local concentration of cetuximab [51]. A recent clinical trial established the safety of selective osmotic blood–brain barrier opening with intra-arterial mannitol infusion followed by intra-arterial infusion of cetuximab [52]. Other cetuximab-based developments include the boronation of the EGFR with antibodies [53] or later the construction of conjugates with cytotoxic platinum derivatives [54]. The boronated cetuximab compounds that were used in experimental boron neutron capture radiation therapy were found to be largely ineffective and the cetuximab-based bio-conjugates used with platinum compounds were also very limited in their efficacy. Instead, smaller peptide–platinum compounds were developed. An observation was made that anti-EGFR antibody treatment augments the effect of radiation [55], but most likely this has to be assumed to be an effect on the antibody-exposed vasculature and thus rather an indirect, tumor cell EGFR-independent effect. Direct use of radiolabeled monoclonal antibodies against EGFR have also been disappointing [56].
Another antibody with different properties, nimotuzumab, was developed into phase III. Nimotuzumab is also a ligand binding blocking antibody against EGFR without intrinsic stimulating activity [57]. It has lower affinity than cetuximab and thus binds more specifically to EGFR overexpressing cells. It has shown promising efficacy in multicenter studies [58] and phase II trials for high-grade glioma [59]. In a multicenter, prospective, open label, two-armed, randomized, phase III trial to test the efficacy of additional intravenous nimotuzumab to standard radiochemotherapy [60] (radiation therapy/temozolomide [RT/TMZ]) for newly diagnosed glioblastoma, it showed a signal of efficacy in patients with residual tumor which were O-6-methylguanine-DNA methyltransferase (MGMT) non-methylated. Unfortunately, the inclusion criteria were very broad and thus the study was not powered to prove such subgroup efficacy [61]. Nimotuzumab is currently used in clinical trials in pediatric brain-stem gliomas [62, 63]. In a clinical trial in newly diagnosed diffuse intrinsic pontine glioma (DIPG) from the University Milano, the combination of radiation plus nimotuzumab plus vinorelbine resulted in a median survival of 15 months [64].
In addition to the effects on receptor activity, purely immunological mechanisms are also conceivable when treating with antibodies by means of ADCC, being a well defined process [65]. The contribution of purely immunologic effector mechanisms to antibody-based treatment of glioblastoma is, however, unresolved and will remain so in the context of human clinical trials for some time as trials do not assess basic humoral or cellular immunologic parameters. The contribution to treatment of glioblastoma in most trial designs is most likely negligible. Even when discussing the mechanism of efficacy of other widely used monoclonal antibodies of extracranial cancer like herceptin, the role for ADCC is somewhat speculative [66] and strategies aiming at enhancing this effect are not clinically used yet [67]. Estimating immunological components like ADCC in gliomas would be pure guesswork in the presence of the well described immunosuppressive environment [68] and there is only one in vitro study report on ADCC in the context of cetuximab [69]. Even if on-treatment biopsies of patients were available to look at lymphocyte content and subtyping, they would probably be wide open to interpretation [70, 71].
3.2.1 Monoclonal Antibody Delivery Issues
As for the issue with antibody treatment of intracranial tumors, the blood–brain barrier as an obstacle to the passage of those large molecules into the brain/tumor parenchyma needs to be considered. However, there is mounting experience with monoclonal antibodies for intracerebral tumors [72, 73]. With bevacizumab, a humanized monoclonal antibody against VEGF to treat malignant glioma, dramatic imaging effects can be achieved [74]. These may be due to neutralizing the intravascular circulating VEGF and thus depriving the angiogenic tumor endothelium of it. The VEGF produced by tumor cells however, being in the interstitial compartment and acting on abluminal receptors, would still be active unless there was some penetration into the tumor, which therefore is assumed to take place. Bevacizumab has also been shown to be a very active agent against radiation necrosis in the brain [75]. The disappointing pivotal trials with bevacizumab are, however, reason to seek improved delivery across the blood–brain barrier by using focused ultrasound for transient barrier opening [76].
Monoclonal antibodies are also considered in the treatment of primary CNS lymphoma [77]. Rituximab, an antibody used for treatment of lymphoma, has been studied in this condition and it could be found in the cerebrospinal fluid after intravenous administration, although only at 0.1% of the systemic concentration, but that nevertheless proves some permeability [78].
3.3 Targeted Toxins
Toxins or conjugates based on antibody-based delivery are large molecules that will not sufficiently cross the blood–brain barrier and have too high a risk for unwanted side effects when administered systemically. Therefore, the need to devise new delivery methods has been recognized and direct intraparenchymal delivery methods are being developed [79] (for review). The paradigmatic method is direct slow catheter-mediated infusion with slow flow rates, which creates a positive pressure wave and can achieve high concentrations of large molecules on the other side of the blood–brain barrier. This so-called ‘convection-enhanced delivery’ (CED) [80] can potentially saturate large tissue volumes . Only one phase III trial using this method has been completed in glioblastoma. In this trial the IL-13 receptor was targeted with a compound in which IL-13 was fused to a truncated pseudomonas exotoxin (cintredikin besudotox) and given by convection over 5 days [81]. Unfortunately this treatment was not superior to its comparator, locally implanted carmustine wafers [82].
CED has also been used for a variety of toxin conjugates based on the EGFR. Targeting the EGFR with toxins is based on the concept of internalization after binding to the receptor (see above). TP-38 is a paradigmatic molecule of that class consisting of TGFα conjugated with a truncated pseudomonas exotoxin similar to the cintredikin besudotox. This has been shown to be safe and effective in a phase I clinical trial where it was also intraparenchymally delivered by convection, but unfortunately it is currently not further developed [41]. More such molecules are, however, under development and await clinical testing [83,84,85,86]
3.4 EGFR as Effector Agent "Address"
3.4.1 CAR-T cells
Immunotherapy has become a major area of promise in the therapy of cancer. A relatively new technology is the engineering of T cells which recognize their target independently from MHC-mediated antigen presentation by expressing a chimeric antigen receptor, so-called Chimeric antigen receptor (CAR)-T cells [87]. This technology has been successfully used in many cancer paradigms, especially leukemia, but the lack of specificity of most antigens, which are also found on normal cells, leads to side effects. Glioblastomas express the already mentioned EGFRvIII, which has in itself a unique site of antigenicity and is a truly specific cancer antigen. It has thus been possible to engineer a CAR that recognizes the vIII receptor mutation via a humanized single-chain antibody variable fragment (scFv), which is fused with the key constituents of the intra-cytoplasmic signaling domains of the T-cell receptor [88]. When expressed in genetically engineered T cells, this leads to specific CAR-T cells against vIII-expressing glioblastoma cells. This is currently under investigation in early clinical trials [89]. Similar reagents have been under preclinical evaluation before [90, 91], and it is expected that there will be a multitude of reagents developed [92]. It is particularly attractive that there is apparent efficacy also against glioma-initiating or glioma stem cells [90].
3.4.2 Nanoparticles
Nanoparticles are vesicular carriers that enable drug release at defined targets while protecting their content from degradation or elimination during transport, and have a long history of pharmacological development also in the context of glioblastoma [93]. Nanocarriers such as liposomes, polymeric nanoparticles, or lipid nanocapsules take advantage of the increasing size of fenestrations in the capillary endothelium of malignant gliomas leading to a partial loss of barrier integrity of the blood–brain tumor barrier [94, 95]. The resulting enhanced permeability and retention (EPR) effect [96, 97] enables large molecules and nanoparticles to become trapped in the interstitium of tumors when barriers become leaky. In addition to this non-specific effect, construction of nanocarriers including targeting antibodies or specific receptor ligands increases the likelihood of selective accumulation in tumors, albeit only in the coherent tumor and not in the area of infiltration. Consequently, the EGFR is a molecule ideal for such tumor targeting in the context of glioblastoma and numerous agents have been constructed.
Extensive preclinical studies have shown promising results, like the use of cetuximab conjugated liposomes, so-called immunoliposomes [98], but there is only limited experience with early clinical trials. Using minicells (400-nm nanoparticles derived from Salmonella typhimurium) that were loaded with doxorubicin and conjugated with panitumumab, EGFR-targeted delivery was evaluated in 14 patients with recurrent glioblastoma [99].
3.4.3 Oncolytic Viruses
Oncolytic viruses have great potential in cancer therapy as they can be designed for many specific characteristics of the respective tumor entities. There are, however, major safety concerns so between design and clinical evaluation, lengthy preclinical studies are mandated. As a result, decades may pass between the description of a promising viral agent and initial clinical trials [100]. Herpes simplex virus (HSV-1)-based constructs are easily manipulated and have already been shown to be safe in phase III clinical trials in melanoma (G207), and received regulatory approval. G47, a third-generation oncolytic HSV-1, is in phase II for glioblastoma [101]. To increase efficacy and safety of oncolytic HSV-1, an EGFR-targeted oncolytic HSV has been progressively developed, by engineering a fully replication-competent virus to specifically infect cells expressing the receptor, which led to highly efficient eradication in a preclinical orthotopic glioma model [102]. However, with persisting safety concerns, this was followed by further virus modification so that by introducing an miR-124 response element into a crucial viral replication gene, the virus is now capable of only reproducing in glioblastoma cells where miR-124 is absent in contrast to the high miR-124 expression in normal glial and neuronal cells of the brain [103]. This agent has promise for clinical trials which are anticipated in the near future.
4 EGFR as an Immunologic Target: Vaccination
EGFRvIII results from a partial gene deletion and this is seen within tumors in a high proportion of patients, albeit usually not in all cells of a tumor [104]. This highlights that intratumoral heterogeneity is a key obstacle to many EGFR-directed therapies. Specific antibodies to the intramolecular unique site of antigenicity have been developed in various immunotherapeutic paradigms [105] as already mentioned. A purely immunological approach to target the EGFRvIII has been attempted by vaccination approaches using the unique antigenic epitope arising within the mutant protein sequence [106]. A synthetic peptide containing that unique amino-acid sequence of the EGFRvIII (rindopepimut) was simply coupled to keyhole limpet hemocyanine and injected intradermally, resulting in an immune response like in a typical vaccination [107]. This therapy has seen very promising phase I and II clinical trials [108], and a specific humoral and cellular immune response was measured in patients. Upon recurrence, 60–80% of the reoperated patients for recurrent tumors showed complete eradication of vIII-positive cells [109]. Unfortunately, however, the pivotal phase III trial for newly diagnosed glioblastoma failed to show overall efficacy[110], with the results still being evaluated for subgroup efficacy.
5 Regulation of EGFR Gene Expression as a Target
In glioblastoma, the complex and interwoven receptor tyrosine kinase (RTK) signaling pathways can be regulated by microRNAs, members of the small non-coding RNA family, by controlling post-transcriptional gene expression by accelerating or blocking the mRNA decay. For example, decreased levels of miR-218 induce the expression of downstream RTK effectors such as phospholipase C-γ1 (PLCγ1) and S6 kinase (S6K1) that counteract the negative feedback loop and therefore increase the activation of both EGFR and platelet-derived growth factor receptor alpha (PDGFRα) [111]. The recent discovery that microRNAs can be found encapsulated in extracellular vesicles (EVs) and subsequently taken up by neighboring cells led to new ways to investigate how cells communicate and modulate gene expression through epigenetic rearrangements [112, 113]. Recent work demonstrated that EVs can carry and transfer EGFR [114] and that cell communication via EVs promotes intratumoral heterogeneity in glioblastoma [115]. In a positive way, miR-1 can interact with Annexin A2 (ANAXA2), one of the major EV proteins, to reduce glioblastoma tumorigenicity [116].
In cancer, with activation of the EGFR pathway like in non-small-cell lung cancer (NSCLC), most of the mutation are concentrated on the kinase domain, while in glioblastoma, mutations are mostly found in the extracellular (EC) domain leading to a poor response to EGFR inhibitors such as erlotinib [117]. In addition, recent work showed that methylation of the EC domain by protein arginine methyltransferase 1 (PRMT1) enhanced EGF binding and therefore dimerization. This led to increased activation of the receptor, counteracting the effect of cetuximab in a mouse model of colon cancer [118]. The expression of the mutant EGFRvIII is also under tight control by epigenetic mechanisms as the inhibition of histone deacetylation resulted in reduction of EGFRvIII expression, which is thought to explain the relatively low and sparse EGFRvIII expression in tumor regions with high EGFR amplification and rearrangement [119, 120]. Epigenetic mechanisms that control EGFR expression were found to be similar in the germinal matrix and gliomas, and it is probable that dysregulation of a locus-specific role for chromatin remodeling in EGFR expression in normal human neural development is involved in gliomagenesis. Strategies targeting these mechanisms have not entered any preclinical development yet [121].
6 General Aspects of EGFR Targeting and Intratumoral Heterogeneity
Many different kinds of EGFR alterations exist simultaneously to a various extent in any glioblastoma (Fig. 1). The complexity of genomic alteration in EGFR and therefore its targeting was recently highlighted by The Cancer Genome Atlas (TCGA). Based on the RNA sequencing analysis of 164 glioblastoma, Brennan and colleagues confirmed that somatic alterations in EGFR regroup into more than EGFR locus amplification associated with exon 2–7 deletion (EGFRvIII), but also exon 12–13 and 14–15 deletion in the extracellular domain as well as C-terminal deletion in exon 25–27 [122]. Additionally, point mutation and protein fusion can also be detected [9] [123]. Most of the minor EGFR variants can be found only in a restricted subpopulation (allelic fraction <10%) and a new single-cell analysis approach based on DNA [124] and RNA [125] sequencing demonstrated that when five alterations in EGFR are found in one tumor resection, up to 32 different subpopulation can be identified, creating a wide range of different targets and potential resistance (Fig. 1) [125]. Moreover, targeting RTK in glioblastoma has been proven difficult due to the co-amplification of multiple RTK (mostly EGFR, PDGFRα, and MET), resulting in intratumoral heterogeneity with many subpopulations with different RTK expression profiles [126, 127]. This is the most likely cause for the disappointment with individual RTK targeting in clinical trials for glioblastoma. As specific subpopulations or clones can be responsible for tumor growth and propagation such as EGFRvIII by enhancing neighboring cell proliferation through IL6 and LIF secretion [128] and activation of the STAT3 pathway [129, 130], treatment with combinatorial RTK inhibitors and STAT signaling pathway might more efficiently target such glioblastoma-driving subpopulations. One aspect to consider in glioblastoma is that compared with other cancer types harboring EGFR amplification, no clonal resistance was observed after EGFR inhibitor treatment. In NSCLC, for example, patients acquire T790M point mutation in exon 20 after erlotinib or gefitinb treatment and therefore develop resistance. No such second-site mutations were discovered in glioblastoma, despite patients developing resistance to EGFR inhibitors. One hypothesis is that the presence of the EGFRvIII mutant increases activation of the PI3K-AKT-mTOR pathway, and when associated with alteration in the tumor suppressor PTEN leads to an increased inhibition of EGFR to achieve the same level of inhibition and overcome resistance [131]. More recently, single-cell analysis of the dynamic evolution of EGFR amplification showed that resistance to EGFR inhibitors in glioblastoma was mostly due to the elimination of the extrachromosomal DNA containing the EGFR amplification (double minutes). Through a highly adaptive process, glioblastoma tumor cells become resistant to lapatinib by losing their EGFR amplified double minutes, which could be shown to re-appear after end of treatment [132].
Despite all these issues, which over the years have shown how difficult EGFR is as a potential target for treating glioblastoma, many reagents are in development after taking these experiences into consideration. Mostly, EGFR is used as an address rather than a single molecule to be knocked out of the signaling pathways (Table 2). Understanding the consequences of EGFR targeting in glioblastoma and the adaptive mechanisms, combinatorial approaches are mandatory [37] and many will include an immunotherapeutic strategy like immune checkpoint inhibition or metabolic approaches [133, 134].
7 Conclusion
Although overexpression of EGFR and mutations of EGFR are one of the most characteristic features of glioblastoma, the cell-biological complexity of this target has outgrown therapeutic developments for decades. While the signaling properties of EGFR and the signaling cascades lose attraction for glioblastoma therapy, EGFR becomes attractive as a docking molecule, which by internalization after ligand binding to the extracellular domain will carry effector agents like monoclonal antibody conjugated toxins or oncolytic viruses into the cells. As EGFR has almost no role in the adult brain, the drug delivery issues which have hampered effective treatment by lack of blood–brain barrier permeability for large effector agents will be addressed by the development and refinement of intraparenchymal delivery methods. Solving the delivery issues of currently developed highly effective and compartmentally non-toxic molecules will eventually close the gap between effective extracranial EGFR targeting and the so-far disappointing attempts at targeting EGFR in glioblastoma.
References
Yewale C, Baradia D, Vhora I, Patil S, Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013;34(34):8690–707.
Downward J, Yarden Y, Mayes E, Scrace G, Totty N, Stockwell P, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307(5951):521–7.
Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313(5998):144–7.
Maire CL, Ligon KL. Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro Oncol. 2014;16(Suppl 8):viii1–6.
Liu F, Hon GC, Villa GR, Turner KM, Ikegami S, Yang H, et al. EGFR mutation promotes glioblastoma through epigenome and transcription factor network remodeling. Mol Cell. 2015;60(2):307–18.
Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA. 1994;91(16):7727–31.
Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25(16):2288–94.
Chen JR, Xu HZ, Yao Y, Qin ZY. Prognostic value of epidermal growth factor receptor amplification and EGFRvIII in glioblastoma: meta-analysis. Acta Neurol Scand. 2015;132(5):310–22.
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110.
Kuan CT, Wikstrand CJ, Bigner DD. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr Relat Cancer. 2001;8(2):83–96.
Padfield E, Ellis HP, Kurian KM. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front Oncol. 2015;5:5.
Zhu JJ, Wong ET. Personalized medicine for glioblastoma: current challenges and future opportunities. Curr Mol Med. 2013;13(3):358–67.
Styren SD, DeKosky ST, Rogers J, Mufson EJ. Epidermal growth factor receptor expression in demented elderly: localization to vascular endothelial cells of brain, pituitary and skin. Brain Res. 1993;615(2):181–90.
Birecree E, King LE Jr, Nanney LB. Epidermal growth factor and its receptor in the developing human nervous system. Brain Res Dev Brain Res. 1991;60(2):145–54.
Wong RW. Transgenic and knock-out mice for deciphering the roles of EGFR ligands. Cell Mol Life Sci. 2003;60(1):113–8.
Wang J, Yu RK. Interaction of ganglioside GD3 with an EGF receptor sustains the self-renewal ability of mouse neural stem cells in vitro. Proc Natl Acad Sci USA. 2013;110(47):19137–42.
Suslov O, Silver DJ, Siebzehnrubl FA, Orjalo A, Ptitsyn A, Steindler DA. Application of an RNA amplification method for reliable single-cell transcriptome analysis. Biotechniques. 2015;59(3):137–48.
Weissleder C, Fung SJ, Wong MW, Barry G, Double KL, Halliday GM, et al. Decline in proliferation and immature neuron markers in the human subependymal zone during aging: relationship to EGF- and FGF-related transcripts. Front Aging Neurosci. 2016;8:274.
Zhou L, Liu J, Xue J, Xu Y, Gong Y, Deng L, et al. Whole brain radiotherapy plus simultaneous in-field boost with image guided intensity-modulated radiotherapy for brain metastases of non-small cell lung cancer. Radiat Oncol. 2014;21(9):117.
Miller JA, Kotecha R, Ahluwalia MS, Mohammadi AM, Chao ST, Barnett GH, et al. Overall survival and the response to radiotherapy among molecular subtypes of breast cancer brain metastases treated with targeted therapies. Cancer. 2017;123(12):2283-93.
Westphal M, Herrmann HD. Epidermal growth factor–receptors on cultured human meningioma cells. Acta Neurochir (Wien). 1986;83(1–2):62–6.
Caltabiano R, Barbagallo GM, Castaing M, Cassenti A, Senetta R, Cassoni P, et al. Prognostic value of EGFR expression in de novo and progressed atypical and anaplastic meningiomas: an immunohistochemical and fluorescence in situ hybridization pilot study. J Neurosurg Sci. 2013;57(2):139–51.
Moazzam AA, Wagle N, Zada G. Recent developments in chemotherapy for meningiomas: a review. Neurosurg Focus. 2013;35(6):E18.
Caraglia M, Marra M, Meo G, Addeo SR, Tagliaferri P, Budillon A. EGF-R small inhibitors and anti-EGF-R antibodies: advantages and limits of a new avenue in anticancer therapy. Recent Pat Anticancer Drug Discov. 2006;1(2):209–22.
Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GH, et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol. 2010;98(1):93–9.
Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol. 2014;16(4):567–78.
Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12(1):95–103.
Raizer JJ, Giglio P, Hu J, Groves M, Merrell R, Conrad C, et al. A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J Neurooncol. 2016;126(1):185–92.
Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys. 2011;80(2):347–53.
Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, et al. RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys. 2013;85(5):1206–11.
Reardon DA, Nabors LB, Mason WP, Perry JR, Shapiro W, Kavan P, et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro Oncol. 2015;17(3):430–9.
Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res. 2013;19(4):900–8.
Iwamoto FM, Lamborn KR, Robins HI, Mehta MP, Chang SM, Butowski NA, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro Oncol. 2010;12(8):855–61.
Yoshida Y, Ozawa T, Yao TW, Shen W, Brown D, Parsa AT, et al. NT113, a pan-ERBB inhibitor with high brain penetrance, inhibits the growth of glioblastoma xenografts with EGFR amplification. Mol Cancer Ther. 2014;13(12):2919–29.
Liu X, Ide JL, Norton I, Marchionni MA, Ebling MC, Wang LY, et al. Molecular imaging of drug transit through the blood-brain barrier with MALDI mass spectrometry imaging. Sci Rep. 2013;04(3):2859.
Lassman AB, Rossi MR, Raizer JJ, Abrey LE, Lieberman FS, Grefe CN, et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res. 2005;11(21):7841–50.
Roth P, Weller M. Challenges to targeting epidermal growth factor receptor in glioblastoma: escape mechanisms and combinatorial treatment strategies. Neuro Oncol. 2014;16(Suppl 8):viii14–9.
Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–90.
Boll W, Gallusser A, Kirchhausen T. Role of the regulatory domain of the EGF-receptor cytoplasmic tail in selective binding of the clathrin-associated complex AP-2. Curr Biol. 1995;5(10):1168–78.
Trowbridge IS, Collawn JF, Hopkins CR. Signal-dependent membrane protein trafficking in the endocytic pathway. Annu Rev Cell Biol. 1993;9:129–61.
Sampson JH, Akabani G, Archer GE, Berger MS, Coleman RE, Friedman AH, et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro Oncol. 2008;10(3):320–9.
Martens T, Laabs Y, Gunther HS, Kemming D, Zhu Z, Witte L, et al. Inhibition of glioblastoma growth in a highly invasive nude mouse model can be achieved by targeting epidermal growth factor receptor but not vascular endothelial growth factor receptor-2. Clin Cancer Res. 2008;14(17):5447–58.
Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W, et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. 2010;65(2):353–61.
Burel-Vandenbos F, Turchi L, Benchetrit M, Fontas E, Pedeutour Z, Rigau V, et al. Cells with intense EGFR staining and a high nuclear to cytoplasmic ratio are specific for infiltrative glioma: a useful marker in neuropathological practice. Neuro Oncol. 2013;15(10):1278–88.
Nicholas MK, Lukas RV, Jafri NF, Faoro L, Salgia R. Epidermal growth factor receptor—mediated signal transduction in the development and therapy of gliomas. Clin Cancer Res. 2006;12(24):7261–70.
Boockvar JA, Kapitonov D, Kapoor G, Schouten J, Counelis GJ, Bogler O, et al. Constitutive EGFR signaling confers a motile phenotype to neural stem cells. Mol Cell Neurosci. 2003;24(4):1116–30.
Hou J, Deng Q, Zhou J, Zou J, Zhang Y, Tan P, et al. CSN6 controls the proliferation and metastasis of glioblastoma by CHIP-mediated degradation of EGFR. Oncogene. 2017;36(8):1134–44.
Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol. 2009;20(9):1596–603.
Hasselbalch B, Lassen U, Hansen S, Holmberg M, Sorensen M, Kosteljanetz M, et al. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro Oncol. 2010;12(5):508–16.
Combs SE, Heeger S, Haselmann R, Edler L, Debus J, Schulz-Ertner D. Treatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (GERT)–phase I/II trial: study protocol. BMC Cancer. 2006;18(6):133.
Hicks MJ, Chiuchiolo MJ, Ballon D, Dyke JP, Aronowitz E, Funato K, et al. Anti-epidermal growth factor receptor gene therapy for glioblastoma. PLoS One. 2016;11(10):e0162978.
Chakraborty S, Filippi CG, Wong T, Ray A, Fralin S, Tsiouris AJ, et al. Superselective intraarterial cerebral infusion of cetuximab after osmotic blood/brain barrier disruption for recurrent malignant glioma: phase I study. J Neurooncol. 2016;128(3):405–15.
Wu G, Yang W, Barth RF, Kawabata S, Swindall M, Bandyopadhyaya AK, et al. Molecular targeting and treatment of an epidermal growth factor receptor-positive glioma using boronated cetuximab. Clin Cancer Res. 2007;13(4):1260–8.
Barth RF, Wu G, Meisen WH, Nakkula RJ, Yang W, Huo T, et al. Design, synthesis, and evaluation of cisplatin-containing EGFR targeting bioconjugates as potential therapeutic agents for brain tumors. Onco Targets Ther. 2016;9:2769–81.
Eller JL, Longo SL, Kyle MM, Bassano D, Hicklin DJ, Canute GW. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery. 2005;56(1):155–62 (discussion 62).
Li L, Quang TS, Gracely EJ, Kim JH, Emrich JG, Yaeger TE, et al. A Phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J Neurosurg. 2010;113(2):192–8.
Talavera A, Friemann R, Gomez-Puerta S, Martinez-Fleites C, Garrido G, Rabasa A, et al. Nimotuzumab, an antitumor antibody that targets the epidermal growth factor receptor, blocks ligand binding while permitting the active receptor conformation. Cancer Res. 2009;69(14):5851–9.
Solomon MT, Selva JC, Figueredo J, Vaquer J, Toledo C, Quintanal N, et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma patients: results from a randomized, double blind trial. BMC Cancer. 2013;13:299.
Bode U, Massimino M, Bach F, Zimmermann M, Khuhlaeva E, Westphal M, et al. Nimotuzumab treatment of malignant gliomas. Expert Opin Biol Ther. 2012;12(12):1649–59.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96.
Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, Mehdorn M, et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur J Cancer. 2015;51(4):522–32.
Massimino M, Bode U, Biassoni V, Fleischhack G. Nimotuzumab for pediatric diffuse intrinsic pontine gliomas. Expert Opin Biol Ther. 2011;11(2):247–56.
Wolff JE, Rytting ME, Vats TS, Zage PE, Ater JL, Woo S, et al. Treatment of recurrent diffuse intrinsic pontine glioma: the MD Anderson Cancer Center experience. J Neurooncol. 2012;106(2):391–7.
Massimino M, Biassoni V, Miceli R, Schiavello E, Warmuth-Metz M, Modena P, et al. Results of nimotuzumab and vinorelbine, radiation and re-irradiation for diffuse pontine glioma in childhood. J Neurooncol. 2014;118(2):305–12.
Herberman RB, Reynolds CW, Ortaldo JR. Mechanism of cytotoxicity by natural killer (NK) cells. Annu Rev Immunol. 1986;4:651–80.
Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol. 2010;28(28):4390–9.
Lu H, Dietsch GN, Matthews MA, Yang Y, Ghanekar S, Inokuma M, et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin Cancer Res. 2012;18(2):499–509.
Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015;17(Suppl 7):vii9–14.
Fukai J, Nishio K, Itakura T, Koizumi F. Antitumor activity of cetuximab against malignant glioma cells overexpressing EGFR deletion mutant variant III. Cancer Sci. 2008;99(10):2062–9.
Donson AM, Birks DK, Schittone SA, Kleinschmidt-DeMasters BK, Sun DY, Hemenway MF, et al. Increased immune gene expression and immune cell infiltration in high-grade astrocytoma distinguish long-term from short-term survivors. J Immunol. 2012;189(4):1920–7.
Yu JS, Lee PK, Ehtesham M, Samoto K, Black KL, Wheeler CJ. Intratumoral T cell subset ratios and Fas ligand expression on brain tumor endothelium. J Neurooncol. 2003;64(1–2):55–61.
Lampson LA. Monoclonal antibodies in neuro-oncology: Getting past the blood-brain barrier. MAbs. 2011;3(2):153–60.
Chen KS, Mitchell DA. Monoclonal antibody therapy for malignant glioma. Adv Exp Med Biol. 2012;746:121–41.
Ellingson BM, Cloughesy TF, Lai A, Nghiemphu PL, Mischel PS, Pope WB. Quantitative volumetric analysis of conventional MRI response in recurrent glioblastoma treated with bevacizumab. Neuro Oncol. 2011;13(4):401–9.
Levin VA, Bidaut L, Hou P, Kumar AJ, Wefel JS, Bekele BN, et al. Randomized double-blind placebo-controlled trial of bevacizumab therapy for radiation necrosis of the central nervous system. Int J Radiat Oncol Biol Phys. 2011;79(5):1487–95.
Chandramohan V, Pegram CN, Piao H, Szafranski SE, Kuan CT, Pastan IH, et al. Production and quality control assessment of a GLP-grade immunotoxin, D2C7-(scdsFv)-PE38KDEL, for a phase I/II clinical trial. Appl Microbiol Biotechnol. 2017;101(7):2747–66.
Wong ET. Management of central nervous system lymphomas using monoclonal antibodies: challenges and opportunities. Clin Cancer Res. 2005;11(19 Pt 2):7151s–7s.
Rubenstein JL, Combs D, Rosenberg J, Levy A, McDermott M, Damon L, et al. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood. 2003;101(2):466–8.
Drapeau A, Fortin D. Chemotherapy delivery strategies to the central nervous system: neither optional nor superfluous. Curr Cancer Drug Targets. 2015;15(9):752–68.
Jahangiri A, Chin AT, Flanigan PM, Chen R, Bankiewicz K, Aghi MK. Convection-enhanced delivery in glioblastoma: a review of preclinical and clinical studies. J Neurosurg. 2017;126(1):191–200.
Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G, et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010;12(8):871–81.
Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5(2):79–88.
Cohen KA, Liu T, Bissonette R, Puri RK, Frankel AE. DAB389EGF fusion protein therapy of refractory glioblastoma multiforme. Curr Pharm Biotechnol. 2003;4(1):39–49.
Chandramohan V, Bao X, Keir ST, Pegram CN, Szafranski SE, Piao H, et al. Construction of an immunotoxin, D2C7-(scdsFv)-PE38KDEL, targeting EGFRwt and EGFRvIII for brain tumor therapy. Clin Cancer Res. 2013;19(17):4717–27.
Chandramohan V, Pegram CN, Piao H, Szafranski SE, Kuan CT, Pastan IH, et al. Production and quality control assessment of a GLP-grade immunotoxin, D2C7-(scdsFv)-PE38KDEL, for a phase I/II clinical trial. Appl Microbiol Biotechnol. 2017;101(7):2747–66
Niesen J, Stein C, Brehm H, Hehmann-Titt G, Fendel R, Melmer G, et al. Novel EGFR-specific immunotoxins based on panitumumab and cetuximab show in vitro and ex vivo activity against different tumor entities. J Cancer Res Clin Oncol. 2015;141(12):2079–95.
Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017;168(4):724–40.
Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7(275):275ra22.
Ruella M, Levine BL. Smart CARS: optimized development of a chimeric antigen receptor (CAR) T cell targeting epidermal growth factor receptor variant III (EGFRvIII) for glioblastoma. Ann Transl Med. 2016;4(1):13.
Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23(10):1043–53.
Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One. 2014;9(4):e94281.
Maus MV, Designing CAR. T cells for glioblastoma. Oncoimmunology. 2015;4(12):e1048956.
Karim R, Palazzo C, Evrard B, Piel G. Nanocarriers for the treatment of glioblastoma multiforme: current state-of-the-art. J Control Release. 2016;10(227):23–37.
Schlageter KE, Molnar P, Lapin GD, Groothuis DR. Microvessel organization and structure in experimental brain tumors: microvessel populations with distinctive structural and functional properties. Microvasc Res. 1999;58(3):312–28.
Brigger I, Morizet J, Aubert G, Chacun H, Terrier-Lacombe MJ, Couvreur P, et al. Poly(ethylene glycol)-coated hexadecylcyanoacrylate nanospheres display a combined effect for brain tumor targeting. J Pharmacol Exp Ther. 2002;303(3):928–36.
Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74(2–3):72–84.
Saha RN, Vasanthakumar S, Bende G, Snehalatha M. Nanoparticulate drug delivery systems for cancer chemotherapy. Mol Membr Biol. 2010;27(7):215–31.
Mortensen JH, Jeppesen M, Pilgaard L, Agger R, Duroux M, Zachar V, et al. Targeted antiepidermal growth factor receptor (cetuximab) immunoliposomes enhance cellular uptake in vitro and exhibit increased accumulation in an intracranial model of glioblastoma multiforme. J Drug Deliv. 2013;2013:209205.
Whittle JR, Lickliter JD, Gan HK, Scott AM, Simes J, Solomon BJ, et al. First in human nanotechnology doxorubicin delivery system to target epidermal growth factor receptors in recurrent glioblastoma. J Clin Neurosci. 2015;22(12):1889–94.
Buijs PR, Verhagen JH, van Eijck CH, van den Hoogen BG. Oncolytic viruses: from bench to bedside with a focus on safety. Hum Vaccin Immunother. 2015;11(7):1573–84.
Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107(10):1373–9.
Uchida H, Marzulli M, Nakano K, Goins WF, Chan J, Hong CS, et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther. 2013;21(3):561–9.
Mazzacurati L, Marzulli M, Reinhart B, Miyagawa Y, Uchida H, Goins WF, et al. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol Ther. 2015;23(1):99–107.
Wikstrand CJ, Reist CJ, Archer GE, Zalutsky MR, Bigner DD. The class III variant of the epidermal growth factor receptor (EGFRvIII): characterization and utilization as an immunotherapeutic target. J Neurovirol. 1998;4(2):148–58.
Kuan CT, Wikstrand CJ, Bigner DD. EGFRvIII as a promising target for antibody-based brain tumor therapy. Brain Tumor Pathol. 2000;17(2):71–8.
Choi BD, Archer GE, Mitchell DA, Heimberger AB, McLendon RE, Bigner DD, et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009;19(4):713–23.
Heimberger AB, Sampson JH. The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin Biol Ther. 2009;9(8):1087–98.
Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. 2015;17(6):854–61.
Swartz AM, Li QJ, Sampson JH. Rindopepimut: a promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy. 2014;6(6):679–90.
Malkki H. Trial Watch: glioblastoma vaccine therapy disappointment in Phase III trial. Nat Rev Neurol. 2016;12(4):190.
Mathew LK, Huangyang P, Mucaj V, Lee SS, Skuli N, Eisinger-Mathason TS, et al. Feedback circuitry between miR-218 repression and RTK activation in glioblastoma. Sci Signal. 2015;8(375):ra42.
Bronisz A, Godlewski J, Chiocca EA. Extracellular vesicles and MicroRNAs: their role in tumorigenicity and therapy for brain tumors. Cell Mol Neurobiol. 2016;36(3):361–76.
Rooj AK, Mineo M, Godlewski J. MicroRNA and extracellular vesicles in glioblastoma: small but powerful. Brain Tumor Pathol. 2016;33(2):77–88.
Read J, Ingram A, Al Saleh HA, Platko K, Gabriel K, Kapoor A, et al. Nuclear transportation of exogenous epidermal growth factor receptor and androgen receptor via extracellular vesicles. Eur J Cancer. 2017;70:62–74.
Ricklefs F, Mineo M, Rooj AK, Nakano I, Charest A, Weissleder R, et al. Extracellular vesicles from high-grade glioma exchange diverse pro-oncogenic signals that maintain intratumoral heterogeneity. Cancer Res. 2016;76(10):2876–81.
Bronisz A, Wang Y, Nowicki MO, Peruzzi P, Ansari KI, Ogawa D, et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014;74(3):738–50.
Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, et al. Differential sensitivity of glioma-versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2(5):458–71.
Liao HW, Hsu JM, Xia W, Wang HL, Wang YN, Chang WC, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest. 2015;125(12):4529–43.
Del Vecchio CA, Giacomini CP, Vogel H, Jensen KC, Florio T, Merlo A, et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene. 2013;32(21):2670–81.
Liffers K, Kolbe K, Westphal M, Lamszus K, Schulte A. Histone deacetylase inhibitors resensitize EGFR/EGFRvIII-overexpressing, erlotinib-resistant glioblastoma cells to tyrosine kinase inhibition. Target Oncol. 2016;11(1):29–40.
Beers R, Chowdhury P, Bigner D, Pastan I. Immunotoxins with increased activity against epidermal growth factor receptor vIII-expressing cells produced by antibody phage display. Clin Cancer Res. 2000;6(7):2835–43.
Cho J, Pastorino S, Zeng Q, Xu X, Johnson W, Vandenberg S, et al. Glioblastoma-derived epidermal growth factor receptor carboxyl-terminal deletion mutants are transforming and are sensitive to EGFR-directed therapies. Cancer Res. 2011;71(24):7587–96.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77.
Francis JM, Zhang CZ, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014;4(8):956–71.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401.
Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell. 2011;20(6):810–7.
Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci USA. 2012;109(8):3041–6.
Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24(16):1731–45.
Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell. 2013;24(4):438–49.
Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, et al. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci USA. 2007;104(31):12867–72.
Vivanco I, Rohle D, Versele M, Iwanami A, Kuga D, Oldrini B, et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc Natl Acad Sci USA. 2010;107(14):6459–64.
Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014;343(6166):72–6.
Gao Q, Lei T, Ye F. Therapeutic targeting of EGFR-activated metabolic pathways in glioblastoma. Expert Opin Investig Drugs. 2013;22(8):1023–40.
Garber ST, Hashimoto Y, Weathers SP, Xiu J, Gatalica Z, Verhaak RG, et al. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol. 2016;18(10):1357–66.
Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel). 2017;9(5):52.
Kwatra MM. A rational approach to target the epidermal growth factor receptor in glioblastoma. Curr Cancer Drug Targets. 2017;17(3):290–6.
Yang W, Barth RF, Wu G, Tjarks W, Binns P, Riley K. Boron neutron capture therapy of EGFR or EGFRvIII positive gliomas using either boronated monoclonal antibodies or epidermal growth factor as molecular targeting agents. Appl Radiat Isot. 2009;67(7–8 Suppl):S328–31.
Acknowledgements
The work in the Laboratory for Brain Tumor Research in the Department of Neurosurgery UKE has received long term support from the Johannes Bauer Foundation, Hamburg and the Rickertsen Stiftung, Hamburg.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The open access fee was paid by the Department of Neurosurgery, University Hospital Hamburg, Hamburg, Germany. No specific funding for this work was received.
Conflict of interest
The authors declare no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Westphal, M., Maire, C.L. & Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 31, 723–735 (2017). https://doi.org/10.1007/s40263-017-0456-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-017-0456-6