
- histopathology
- kaposiform hemangioendothelioma
- bone
- vascular tumors
Kaposiform hemangioendothelioma (KHE) is a rare locally aggressive vascular tumor that mainly occurs in children and early adolescents. It is often associated with the Kasabach–Meritt phenomenon which is marked by severe thrombocytopenia and a variable degree of anemia. The tumor mostly is located in the superficial or deep soft tissue mass of the extremities. Herein, we report an unusual case of kaposiform hemangioendothelioma that, for the first time, was found in the proximal medullary cavity of the tibia and fibula of an 8-year-old boy. This patient was first diagnosed as fibrous dysplasia. He underwent wide local excision, but the mcroscopic examination of the mass was confirmed to be kaposiform haemangioendothelioma (KHE) based on morphological and immunohistochemical findings, which were characterized by having features common to capillary hemangioma and Kaposi sarcoma.
Case Report
This case represents the first report of this disease occurring in the proximal medullary cavity of the tibia and fibula. An otherwise healthy 8-year-old boy felt pain in his left leg. He was not easily bruised or subject to bleeding. The discomfort slowly aggravated over the months. The boy had been born to healthy parents at 40 weeks of gestation by normal delivery. He had no intrauterine disease or perinatal problems. He was vaccinated as scheduled, did not undergo any surgery, and was not admitted for other diseases.
On physical examination, the child was afebrile without abnormal vital signs or weight loss. No tints of blue, pink, or red on his left leg could be seen and no lymph nodes were palpable. The amount of knee extension was 170 degrees and the amount of knee flexion was 65 degrees, both were limited. Tenderness could be felt by the boy when he was deeply pricked. Laboratory results showed the following: alkaline phosphatase (42, 36~213 U/L), normal blood cell counts, hemoglobin (123 g/L, 120~160 g/L), erythrocytes (4.44×1012/L, 4.0~5.5×1012/L), leukocytes (9.20× 109/L, 4.0~10×109/L) and platelets (273×1012/L, 100~300×1012/L). A conventional X-ray examination of the proximal tibia and fibula (Fig.1) showed a large, centrally located, lytic lesion in the metaphyseal-epiphyseal area with thinning of the surrounding cortex, minimal dilatation and fairly sharp margination that would suggest a benign disease, such as fibrous dysplasia or perhaps a giant cell tumor. In addition, there was minimal evidence of matrix calcification and very little evidence of a periosteal response in the surrounding bone.
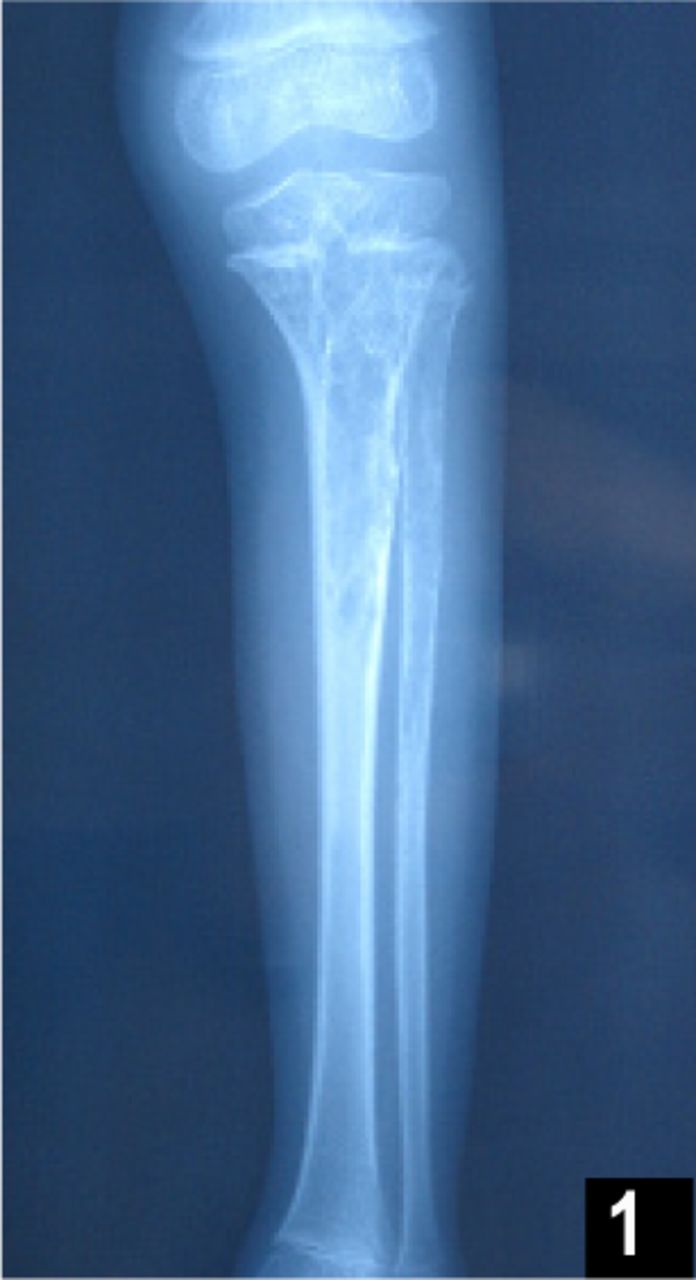
This shows a large, centrally located, lytic lesion in the tibial and fibula metaphyseal-epiphyseal area with thinning of the surrounding cortex, minimal dilatation and fairly sharp margination.
The patient was first diagnosed as having fibrous dysplasia and underwent wide local excision without any problems or complications in our hospital. During the operation, a cyst containing a dark translucent liquid was detected in the proximal medullary cavity of the tibia and fibula. The surrounding cortex was as thin as 1 mm and even the medullary cavity could join with the knee joint due to the expansion of the cyst. Allograft bony pieces mixed with bone morphogenetic proteins and gentamicin were planted in the cavity after the excision. During the operation an anatomical AO steel plate was used for support. Perioperative fluid therapy and antibiotic therapy were prescribed. Transfusion was not used. A conventional X-ray examination after operation is shown in Fig.2.
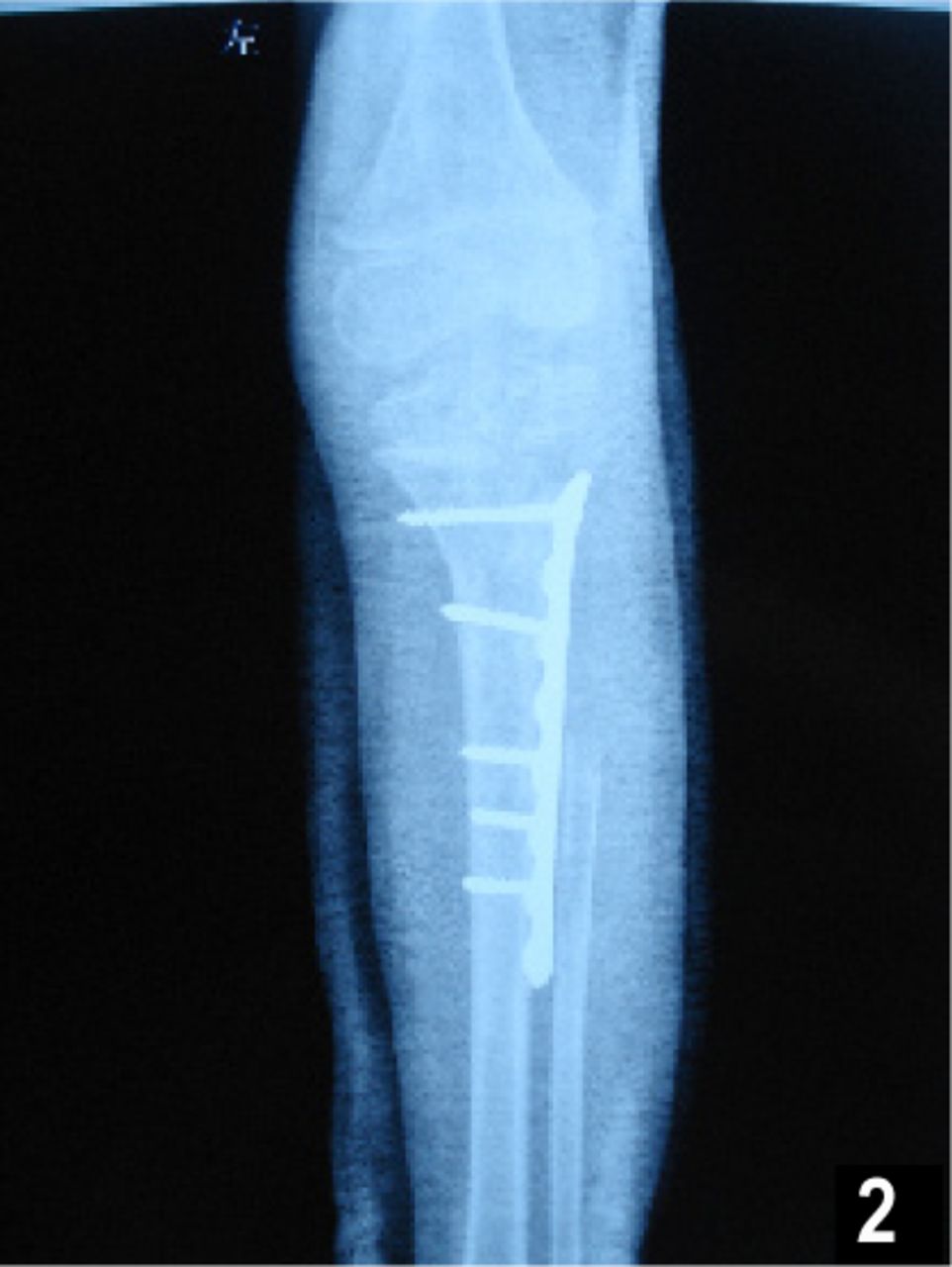
This shows autogeneous bony pieces were planted in the cavity after the excision. During the operation an anatomical AO steel plate was used for support.
Microscopic examination of the mass confirmed the lesion to be kaposiform haemangioendothelioma (KHE) based on morphological and immunohistochemical findings. Microscopically, the tumour consisted of “glomeruloid” solid nests interspersed with capillaries. The “glomeruloid” nests were made up of spindle and round cells, with apparent slit-like spaces. Atypia and mitosis were minimal. Immunohistochemical stains showed positive expression for CD34 (Fig. 3A), CD31, Factor VIII and HHF35, but negative for expression of Des. Heamosiderin deposition was obvious (Fig.3B). Many alpha-smooth muscle actin-positive spindle cells were found around the vascular spaces. The morphologic picture was consistent with the aggressive growth of a hemangioendothelioma, most likely of the kaposiform type. Differential diagnosis included osteofibrous dysplasia, Kaposi’s sarcoma and capillary hemangioma. Although the representation in the X-ray suggested a diagnosis of osteofibrous dysplasia, it was easily excluded under microscopic examination. Intracellular and extracellular hyaline globules and densely packed endothelial cells positive for CD34 forming elongated or crescentic vascular spaces can be seen in both KHE and Kaposi’s sarcoma (KS), but the latter often has a profound peripheral inflammatory infiltrate which was not seen in our case, and variation in nodules from one area to another is much greater in KS. Some nodules is KS possess vessels with round or oval lumens with strong similarity to capillary hemangiomas, but unlike capillary hemangiomas are composed of discrete lobules of small vessels. KHE consists of irregular nodules of tumor that infiltrate the soft tissue[1].
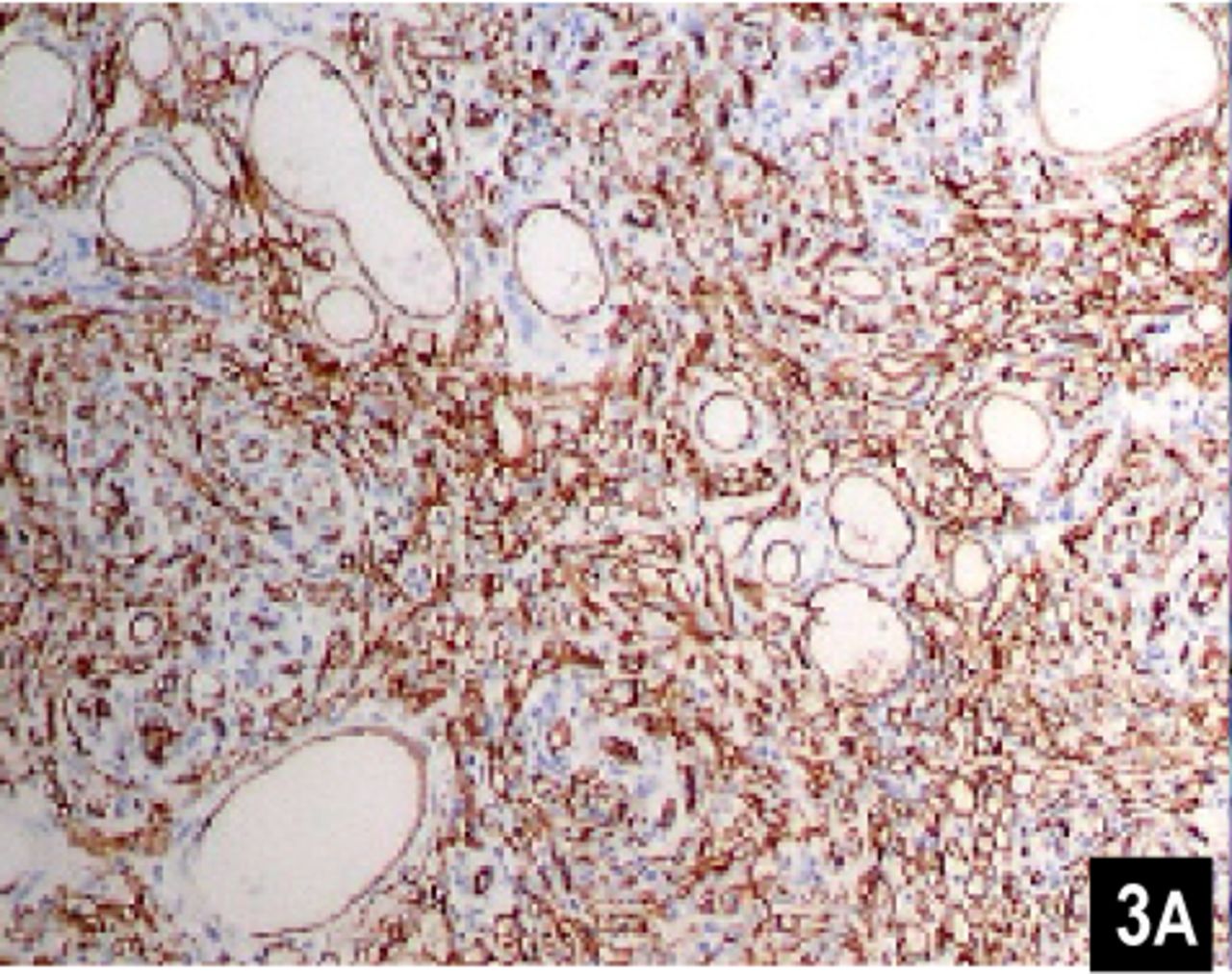
Immunohistochemical examination shows nodules with vessels with round or oval lumens with strong similarity to capillary hemangiomas. CD34 shows a diffuse staining of the lobules, including spindled cells at the periphery and small vascular channels in the center (×100).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
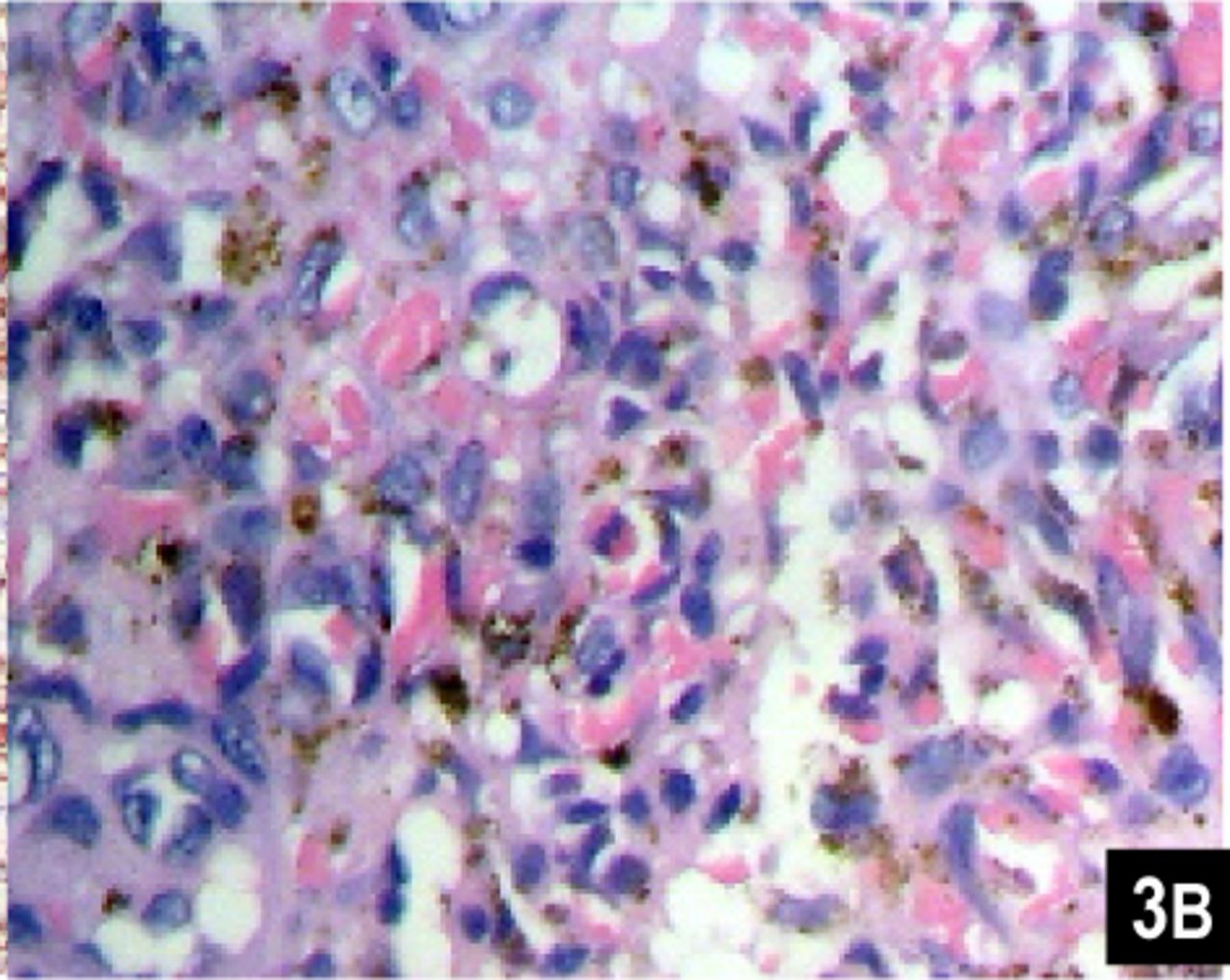
This slide shows round and epithelioid endothelial cells with abundant eosinophilic cytoplasm forming slit-like structures filled with red cells (H&E ×400).
Based on the above analyses, a diagnosis of KHE was made without Kasabach–Merritt phenomenon. With 6 months of follow-up, the patient has been free of disease, and the function of the left knee was preserved. To the best of our knowledge, this unusual case is the first report of kaposiform hemangioendothelioma in the marrow cavity. In contrast to cases that were reported mostly in the soft tissues of the extremities, head and neck, trunk, and body cavity, this case was found in bone.
Review of the Literature
Kaposiform hemangioendothelioma (KHE) was first described in 1993 by Zukerberg et al. 2 as a distinctive vascular neoplasm affecting predominantly children and neonates. It is composed of infiltrating nodules with slit-like or crescentic vessels that are poorly canalized and lined by spindled endothelium cells. Dilated hyperplastic lymphatic channels are sometimes seen which has been called “lymphangiomatosis[2].”
Clinically, KHE is typically an ill-defined, red to purple indurated plaque frequently complicated by Kasabach-Merritt phenomenon (KMP), and looks quite different from infantile hemangioma[3], with little tendency to involute spontaneously[4]. It often occurs in the first decade of life, and manifests later than infantile hemangioma[5]. KHE is considered to be a borderline tumor with a locally aggressive behavior[2,6]. Although there were have been cases of regional lymph node metastasis, to date distant metastasis have not been reported[2,4]. It occurs predominantly in infancy and early childhood without any sex preponderance[2,4,7]. The incidence is very rare, and the mortality rate ranges from a frequency of 12% in 153 cases collected from the world literature up to 24% in a separate smaller series[8,9].
KHE is considered to occur most commonly in the superficial or deep soft tissue mass of the extremities followed by the head and neck[4]. KHE occurring in adults is more rare, and is mainly situated in the superficial soft tissue[10]. The diameter of the tumor ranges from a few millimeters to 50 cm[11,12]. Magnetic resonance images always show an enhancing, ill-defined, soft tissue mass in patients, but no characteristic images can be found to differentiate KHE from other soft-tissue tumors[3,4]. The cases reported involved deep soft tissues of the extremity, body cavity, head and neck, but it has never been reported in the medullary cavity as was found in our case[2,4].
Histologically, KHE has a characteristic appearance. Round and epithelioid endothelial cells with abundant eosinophilic cytoplasm, vessels with round to oval lumens similar to capillary hemangiomas, coexist with spindled neoplastic endothelial cells that form elongated or crescentic vascular spaces similar to Kaposi sarcomas[1,2,4,13]. Tightly coiled and highly convoluted vessels bud directly off of larger vessels, which often appear as crescentic profiles surrounding the smaller vessels depending on the plane of section[4]. Scattered throughout the tumors are epithelioid or glomeruloid areas in which the cells appear distinctly rounded with hyaline granules, finely stippled hemosiderin, and small vacuoles. Central platelet-rich fibrin microthrombi are typically identified in these areas. Approximately two thirds of KHE exhibit lymphatic abnormalities consisting of thin-walled vessels surrounding the vascular tumor nodules, and often extend outward for a considerable distance[4]. Immunohistochemicaly, the predominant neoplastic component of KHE recapitulates the phenotype of blood vascular endothelium (CD31 and CD34 positive) and totally lacks immunoreactivity for GLUT1 and LeY[4,14]. VEGFR-3[15] and D2-40[16,17]are markers recently used for KHEs which are partially lymphothelial differentiated, and can be used to differentiate KHE from other vascular lesions of infancy[16,17].
KHE can occur with or without Kasabach-Merritt phenomenon[18](KMP), which is marked by severe thrombocytopenia and a variable degree of anemia. Most of the KHEs present with KMP, namely co-agulopathic KHE, and cause significant mortality by a deadly haemorrhage[2,4,9]. On the other hand, at least 90% of KMP are secondary to KHE[9, 13,19]. The common association with Kasabach-Merritt phenomenon probably relates in part to unique architectural features that favor turbulent blood flow and platelet activation. Small convoluted capillaries arise directly from large vessels in a serial or linear fashion, arguably creating a situation that results in turbulence leading to platelet activation and aggregation[4]. Kasabach et al.[18] reported that KHE in the extremities and the retroperitoneum are more likely to be associated with KMP. There are fewer KHEs without Kasabach-Merritt phenomenon as in our case, namely noncoagulopathic KHE, which cause a much lower mortality[4,9,19]. These noncoagulopathic KHEs are similar with regard to the sex ratio, age of onset, and appearance to those with coagulopathic KMP[19]. But the noncoagulopathic lesions tended to be more focal and are more likely to be small and superficia[13], and there are also some differences in location in noncoagulopathic KHE as compared with KHE-associated KMP. None of these tumors were located in the retroperitoneum or mediastinum or invaded visceral organs, where outcome was known to be poor[2,9,20]. The reason maybe that KHE arising in these areas can not appear clinically evident unless KMP occurs. In our case, not only the platelet count was normal, but also the thrombin time and activated partial thromboplastin time were within the norm, no Kasabach-Merritt symptoms were apparent, so it belonged to the noncoagulopathic KHE. The prognosis of adults is better. After wide local excision, some patients remain alive with residual disease including KMP[4].
At the present time, the etiology of KHE is unclear, but it has no known association with Kaposi sarcoma related to human immunodeficiency virus infection. The absence of HHV-8 LNA-1 in KHE underscores a different pathogenesis from Kaposi sarcoma[21]. But the concurrence of features of tufted angioma (TA) and KHE in the same specimen of some patients raises the question as to whether these tumors exist on a continuum[12,22]. There are also reports about a dynamic transformation between both tumors within a single patient, which suggests they are two variants of the same vascular tumor[23]. TA is another vascular tumor manifesting as deep red-purple coalescent papules and plaques with ill-defined borders at birth or in fancy and childhood. It can also manifest profound thrombocytopenia, although most lesions are not coagulopathic[1,24]. In fact, microscopic differentiation between KHE and TA can be difficult on a small specimen, and pathologists often have differing interpretations. The spindled cells seen in KHE are occasionally observed in TA, and the crescent-like peripheral clefts seen in TA also have been noted in KHE[2,19,23]. Enjolras et al.[25] noted that TA was more commonly seen with the histologic pattern in early Kasabach-Merritt phenomenon (KMP) or residual lesions, whereas the appearance of KHE was more prominent during active disease. Thus, it is possible that KHE and TA are part of the same neoplastic spectrum[9,23]. Arai et al.[17] reported D2-40 could be a useful antibody for immunohistochemical discrimination between KHE and TA because of the different immunostaining patterns. These differences were limited to the peripheral area of capillary proliferation and surrounding dilated vessels; therefore, it is suggested that KHE and TA may reflect different stages in the evolution of a single entity. Namely, they may originate from stem cells possessing the characteristics of both lymphatic and blood vessel endothelial lineages.
Treatment experience of KHE is limited by its rarity. Although some reports have suggested that KHE can involute spontaneously[3], Lyons et al.[4] insisted that it would not regress in the absence of therapy, and the most effective treatment of KHE is complete surgical excision. Tumors limited to the superficial soft tissues are best treated by wide local excision with clear surgical margins. Masses located in the retroperitoneum are typically extensive, unresectable lesions associated with KMP and frequently lead to patient death[1].
KHE appears with a wide spectrum of behavior and response to pharmacological treatment, and although it can stabilize over time, the tumor never totally regresses, even with successful pharmacologic therapy[25]. Management of KMP has been proved to be a most problematic issue in KHE, and a multimodality approach using steroids, interferon, and cytotoxic agents may be required[26,27]. The two most commonly used medical therapies are steroids and interferon[28]. But treatment with cortisone achieved success in only 10% of all cases, and treatment with interferon alpha in only 50~60%[9,28]. A multimodal intervention including vascular embolization, systemic interferon, cyclophosphamide, epsilon aminocaproic acid, and compression therapy was reported to result in complete resolution of advanced KHE in a 14-month-old patient[29].
Because of its rarity there are no known treatment guidelines for KHE, so when formulating a treatment plan, the risk of coagulopathy with increasing size and possible sequelae of persistent tumor, such as muscle fibrosis and joint contracture should be considered. In addition, Gruman et al.[3] considered KHE with a normal platelet count, limited extent, and without visceral involvement, could be safely followed-up without treatment. Therefore, the decision as to whether or not to treat a noncoagulopathic KHE should be based on the size and location of the tumor and the possible side effects of therapy[18].
- Received April 19, 2007.
- Accepted May 21, 2007.
- Copyright © 2007 by Tianjin Medical University Cancer Institute & Hospital and Springer