

Abstract
OBJECTIVE The purpose of our study was to investigate the expression level of MTA1 mRNA in breast cancer and its significance in relation to clinical pathology.
METHODS The expression levels of MTA1 mRNA in tumor and in paired normal adjacent tissue of 56 cases with breast cancer were detected by fluorescent quantitative polymerase chain reaction.
RESULTS The expression of MTA1 mRNA was detected in 47 tumor specimens of 56 breast cancer patients (83.9%) and was significantly higher than in the paired normal breast tissue. The over expressed MTA1 mRNA was significantly associated with pathologic stage (P = 0.029), clinical grade (P = 0.035) and lymph node status (P = 0.001).
CONCLUSION The over expression of MTA1 mRNA may play a crucial role in the development of breast cancer. As the MTA1 was comparatively highly-expressed in breast cancer, it may become a new biomarker for the diagnosis and treatment of breast cancer in the future.
keywords
- breast cancer
- gene expression
- metastasis-associated gene 1
- fluorescent quantitative polymerase chain reaction
Introduction
The genesis and development of a tumor is an evolving process with multiple phases and is under regulation by many genes. MTA1 (metastasis-associated gene 1), reported in recent years, is closely associated with the metastasis of tumors. Recent research has found that MTA1 plays an important role in the genesis and development of lung cancer, esophageal carcinoma, hepatocellular carcinoma, ovarian cancer, prostatic carcinoma and so on[1-5]. At present, research about the quantitative detection of MTA1 gene in breast cancer has not been reported. In this study we used a very accurate method, fluorescent quantitative polymerase chain reaction (FQ-PCR), to monitor the real time expression of MTA1 mRNA in different stages of breast cancer, and to further investigate the important role of MTA1 in the invasion and metastasis of breast cancer.
Materials and Methods
Clinical data
After the informed consent form was signed, tumor specimens were collected from 56 female breast cancer patients treated at the First Affiliated Hospital of Zhengzhou University during the period of September 2005-March 2006. The median age of the patients was 42 ± 13 years (ranging from 27-75 years). All tumors were diagnosed histologically as invasive breast cancer: invasive ductal carcinoma in 39, invasive lobular carcinoma in 14 and medullary carcinoma in 3. The detailed characteristics of the patients, including age, tumor size, menopausal status, pathologic grade, the presence of lymph node metastasis, and the hormone receptor status, are demonstrated in Table 3. Pathologic grade was determined according to the modified Bloom-Richardson criteria[6]: low-grade (well and moderately differentiated) in 30 cases and high-grade (poorly differentiated) in 26 cases. Among these patients, 16 patients were in stage I, 24 in stage II, 7 in stage III, and 9 in stage IV. The lymph node status of the patients was assessed by histologic examination. ER expression was evaluated using immunohistochemical staining (positive when > 15% of the nuclei showed staining). Twenty one cases were found ER negative, 37.5%.
Breast tissue samples, 0.5-1.0 cm in diameter, were obtained from the fresh specimens at the time of surgical treatment and were characterized pathologically by their stage. The samples were frozen in liquid nitrogen and were kept frozen until they were used. The paired normal breast tissue was obtained from the periphery of each specimens.
Extraction of total RNA and synthesis of cDNA
Total RNA was isolated from tumor tissues and paired adjacent normal tissues using the Trizol Reagent Kit (Promega) and the processing procedures were based on the manufacturer’s instructions. Electrophoresis was performed on the extract in 1% agarose gel and displayed 3 distinct bands of 28 s, 18 s, and 53 s. The UV-1601 spectrophotometer was used to detect the concentration and purity of the RNA. Reverse transcription of cDNA was performed using the AMV Reverse Transcription Reagent Kit (Promega). Sterile DEPC water was used as a blank control for all samples. The verified stock solution of cDNA was preserved under -70°C.
The principle of FQ-PCR
Adding fluorophore to the PCR reaction system and using the accumulation of fluorescent signal for real time monitoring, FQ-PCR can be utilized for quantitative analysis of an unknown template using a standard curve.
The primer design
Primer 5 software was used to design the gene primer of MTA1 and β-actin which was synthesized by the Biotechnology Company of Shanghai. MTA1 forward and reverse primer sequences used were 5’-TGC CCG AAG CCT CCC AGA-3’ and 5’-GTT GCC GTC CAC CCC GTT-3’. β-actin forward and reverse primer sequences were 5’-GAT CAT TGC TCC TCC TGA GC-3’ and 5’-ACT CCT GCT TGC TGA TCC AC-3’.
The detection of reverse transcription
β-actin was used as an internal control for all samples. cDNA that was diluted by 1:20 was used for the PCR reaction. The PCR master mix was as follows: buffer (10 ×) 2 μl, dNTPs (2.5 mM) 1.6 μl, primer 1 (10 μM) of β-actin 0.6 μl, primer 2 (10 μM) of β-actin 0.6 μl, r-Taq enzyme 0.1 μl, cDNA 5 μl, sterile water 10.1 μl. The mixture was initially denatured at 95°C for 2 min followed by 32 cycles of denaturation at 95°C for 20 s, annealing was at 57°C for 20 s and extension at 72°C for 20 s. This was followed by a final cycle at 72°C for 5 min for extension and then the temperature was kept at 4°C. The sterile DEPC water was used as blank control for all samples. The β-actin PCR product was assessed by electrophoresis in agarose gel at 4 V/min for 40 min. The cDNA from reverse transcription was considered valid if the short fragment (100 bp) of β-actin was able to be amplified.
Generation of the standard curve
After the detection of the purity of the PCR product, 1 ng/20 μl was used as the standard concentration for preparation of serial dilutions of 1 × 10-4, 1 × 10-5, 1 × 10-6, 1 × 10-7, 1 × 10-8 to be used in the real-time PCR. Utilizing this, the standard curve could then be generated (Fig. 1).

The standard curve of FQ-PCR of MTA1 mRNA.
The real-time PCR detection of MTA1 in the cDNA
The amplification reaction was performed using Light-Cycler-DNA Master SYBR Green I and the iCycler iQ system (Table 1).
Reaction system of real-time PCR.
The reaction conditions of β-actin were as follows: Cycle1 (1 ×) at 95°C for 2 h and 30 s, Cycle 2 (40 ×) at 95°C for 20 min followed by 57°C for 20 min, 72°C for 20 min and 86°C for 15 min, Cycle 3 (1 ×) at 95°C for 1 h, Cycle 4 (1 ×) at 55°C for 30 min, Cycle 5 (80 ×) at 55°C for 10 min, Cycle 6 (1 ×) preserved at 4°C.
The reaction conditions of MTA1 were as follows: Cycle1 at 95°C for 3h, Cycle 2 (40 ×) at 95°C for 15 min followed by 57°C for 20 min, then 72°C for 18min and then 86°C for 17 min, Cycle 3 (1 ×) at 72°C for 2 h, Cycle 4 (1 ×) at 95°C for 1 h, Cycle 5 (1 ×) at 55°C for 1 h, Cycle 6 (80 ×) at 55°C for 10 min, Cycle 7 (1 ×) preserved at 4°C.
Statistical analysis
SPSS12.0 software was used. The statistical data were analyzed by the Chi-square test and the t-test. The value of P < 0.05 was regarded as statistically significant.
Results
Amplification map and MTA1 dissociation curve
The standard curve, amplification curve and dissociation curve for the real-time PCR of MTA1 showed that the amplification curve of each sample was well. The data were reliable because the Ct value of each sample was above the standard curve. The dissociation curve showed that the amplification of dimeride was not obvious (Figs. 2, 3).

Amplification map of MTA1 gene.

Dissociation curve of MTA1 gene.
Expression of MTA1 genes in breast cancer and paired normal adjacent breast tissue
The present research found that the expression of the MTA1 gene in breast cancer was 83.9% (47/56) which was higher than that in the matched normal breast tissue (Fig. 4) and the difference between the two groups was significant as confirmed by the t-test (P = 0.032, Table 2).
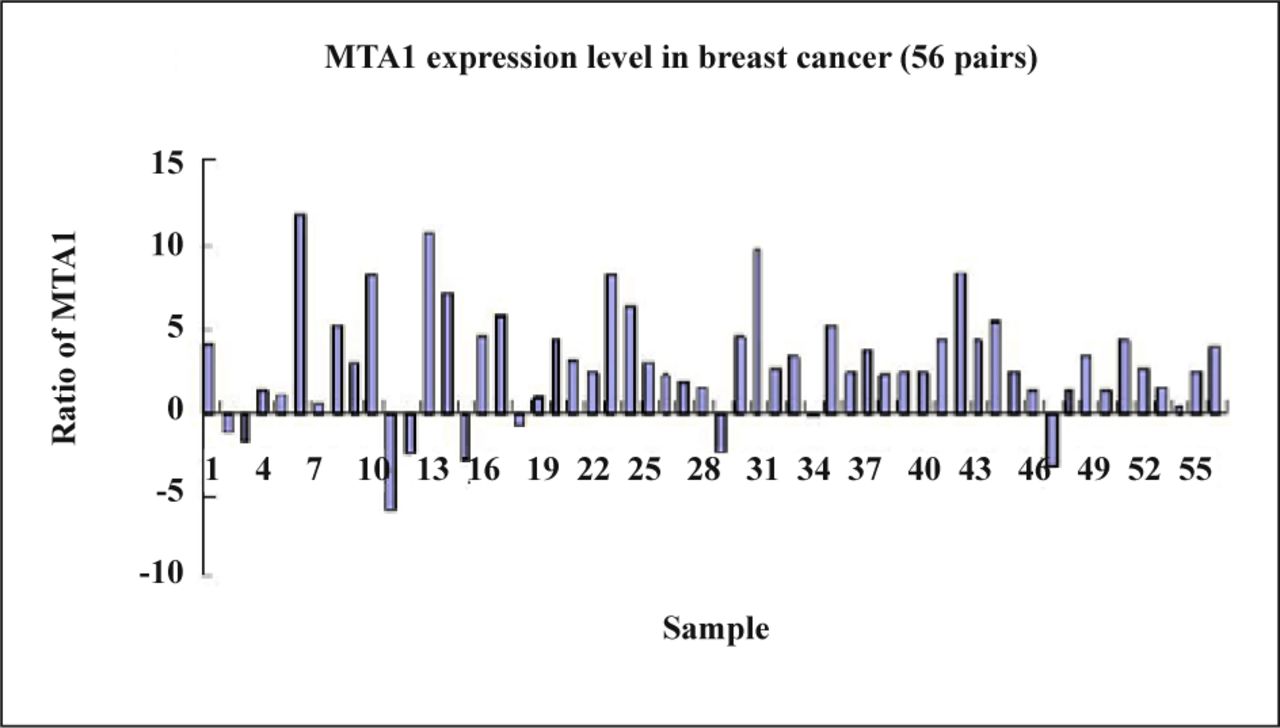
The expression rate of MTA1 gene in the matched normal breast tissues.
Expression of MTA1 genes in breast cancer and matched normal breast tissues.
Relationship between the expression level of MTA1 in breast cancer and the clinical-pathologic features of individuals
The present research found that the expression of MTA1 mRNA had no correlation with the age of patients or with the tumor site. The expression of MTA1 mRNA in breast cancer with lymph node metastasis was significantly higher than in the breast cancer without lymph node metastasis. The difference between the two groups was significant (P = 0.001). With the increase of the tumor size, clinical stage and pathologic grade, the expression level of MTA1 gene was significant, yet the increase was gradual (P = 0.042, P = 0.035, P = 0.029, Table 3).
Relationship between the level of MTA1 mRNA in breast cancer and various clinicalpathologjc features in 56 cases with breast cancer.
Discussion
MTA1 is a novel gene cloned from the rat 13762NF mammary adenocarcinoma metastatic system using the differential hybridization method by Pencil et al.[7] It is called MTA1 (metastasis-associated gene 1) because it has a positive correlation with tumor metastastic potential. The human MTA1 gene is located at chromosome 14q32.3 and encodes a protein of 715 amino acid residues with a molecular mass of 82 kD. It contains 9, 2 and 7 phosphorylation sites for protein kinase C, tyrosine kinase and casein kinase-II, respectively and 4 N-glycosylation sites. Studies indicate that the MTA1 protein may be involved in interactions with other proteins that are essential to normal cellular functions in the signal transduction pathway[8]. A proline-rich stretch at the carboxyl-terminal extremity of the MTA1 protein at residues 696-705 (LPPRPPPPAP) completely matched the consensus sequences for the Src homology 3 domain binding site. Meanwhile Src homology 3 domains are considered to be involved in protein-protein interactions in signal transduction pathways and to regulate some genes associated with invasion and metastasis of tumors[1]. The precise role of the MTA1 protein in the progression of malignant tumors remains unknown. Many studies have emphasized that MTA1 is a subunit of NuRD (nucleosome remodeling and histone deacetylation). It takes part in the formation of histone deacetylase which contains nucleosome remodeling activity. The deacetylation of histone turns some loose domains of chromatin into close ones, and cut off the transcription of some genes, reducing their transcriptional level to enhance invasion and tumor metastasis[2]. Some research has found that MTA1 plays an important role in the genesis and development of lung cancer, esophageal carcinoma, hepatocellular carcinoma, ovarian cancer, and prostatic carcinoma among others[3-5,9,10].
FQ-PCR is a new quantitative detection and analytical technique for nucleic acids. Through FQ-PCR, real time monitoring of the reaction process is possible by adding fluorescent material to the amplification reaction. This has gradually become an important tool in molecular biology research because of the precise quantitation, the high sensitivity, the rapid speed of the reaction, the good reproducibility as well as the fact that electrophoresis is not necessary after the reaction. FQ-PCR can precisely quantify the amount of the initial sample nucleic acid through the generation of a standard curve. In this study, we used FQ-PCR to detect the expression of MTA1 in the 56 cases with breast cancer and in paired normal breast tissue. The results showed that the expression of the MTA1 gene in breast cancer was 83.9% which was significantly higher than that in the matched normal tissues (P = 0.032).
In our further analysis, we found that the expression of MTA1 mRNA had no correlation with the age of the patients or with the tumor site, and that its expression in breast cancer with lymph node metastasis was significantly higher than in the breast cancer without lymph node metastasis (P = 0.001). The expression of MTA1 mRNA in the breast cancer classified as stage III-IV was significantly higher than in the breast cancer classified as stage I-II (P = 0.035). With the increase of the tumor size, the expression of MTA1 gene gradually increased. A significant difference was found among the groups of different tumor size (P = 0.042). This data coincides with the results reported by Toh et al.[4] who detected the expression of MTA1 mRNA in 47 cases with esophageal squamous cell carcinoma using semi-quantitative RT-PCR. Since the quantitation of the initial template in FQ-PCR is more precise, the results of the present study are more reliable.
Furthermore, with advancement of clinical stage, the expression of MTA1 gene gradually increased. The results showed that the expression of MTA1 mRNA in the breast cancer classified as stage III was significantly higher than that in breast cancer classified as stage II and I (P = 0.029). The data coincide with the results reported by Sasak et al.[3] who detected the expression of MTA1 gene in 74 cases with non small cell lung cancer using RT-PCR. All these data show that there is a close correlation between the expression of MTA1 and the clinical stage, pathologic grade, tumor size and lymph node metastasis. The specific expression of the MTA1 gene in breast cancer is higher than that in normal breast tissue. With the increase in tumor size, clinical stage and pathologic stage, the expression of MTA1 mRNA gradually increased. This showed that the MTA1 gene plays an important role in the genesis and evolution of breast cancer in many phases especially in invasion and metastasis. With the previous study, we proposed that the molecular mechanism of MTA1 in the malignant transformation and metastasis of breast cancer cells possibly correlates with the deacetylation of histones. Histones and chromosome can regulate the on-off mechanism of some genes in the process of cell growth, multiplication and differentiation. The abnormal up- or down-regulation of some genes will result in the malignant transformation of cells and possibly play a part in the complicated reticular system regulation of transcription such as the decrease in cell adhesive ability, the expression of adhesion molecules and cytoskeletal proteins and the formation of neovasculature and lymphatic vessels.
Based on this data, we consider that high expression of MTA1 mRNA is an important event in the processes of invasion and metastasis in breast cancer. The MTA1 gene has become a novel target to be investigated for the mechanism of the genesis and development of malignant tumors and for the study of effective ways to diagnose and treat cancer. Further research will provide targeted therapies against malignant tumors and new ideas and perspective, which might create new modes of cancer treatment.
- Received June 9, 2009.
- Accepted August 3, 2009.
- Copyright © 2009 by Tianjin Medical University Cancer Institute & Hospital and Springer
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.