

Abstract
OBJECTIVE To investigate the anti-proliferative effect of rosiglitazone and its relationship to peroxisome proliferatoractivated receptor γ (PPARγ) in human breast cancer cell line MDA-MB-231 and evaluate the potential application value of rosiglitazone for breast cancer therapy.
METHODS The cytostatic effect of rosiglitazone on MDA-MB-231 cells was measured by the MTT assay. Cell-cycle kinetics was assessed by flow cytometry. Apoptotic cells were determined by the TUNEL assay. MDA-MB-231 cells were treated with rosiglitazone or in combination with the PPARγ antagonist GW9662 to investigate the effect of rosiglitazone on cell proliferation and its relationship to PPARγ.
RESULTS The results showed that rosiglitazone could inhibit growth of MDA-MB-231 cells in a dose- and time-dependent manner with an IC50 value of 5.2 μmol/L at 24 h after the drug was added into the culture. Cell cycle analysis showed that the percentage of G0/G1 phase cells increased, S phase cells decreased, and cells were arrested in G1 phase with increasing concentrations of rosiglitazone. Detectable signs of apoptotic cell death caused by rosiglitazone occurred at a concentration of 100 μmol/L and the apoptotic rate was (18 ± 3)%. PPARγ selective antagonist GW9662 could partially reverse the inhibitory effect of rosiglitazone on proliferation of MDA-MB-231 cells.
CONCLUSION It was concluded that rosiglitazone can inhibit growth of MDA-MB-231 cells via PPARγ activation and a high concentration of rosiglitazone can also induce MDA-MB-231 cell apoptosis. These results suggest that PPARγ represents a putative molecular target for chemopreventive therapy and rosiglitazone may be effective in the treatment of breast cancer.
keywords
- peroxisome proliferator-activated receptor γ (PPARγ)
- rosiglitazone
- MDA-MB-231 cells
- antiproliferative effects
- apoptosis KOLLA anti-proliferative
Introduction
Breast cancer is among the leading chronic conditions affecting adult women, with considerable resources devoted to research and disease control efforts, notably screening. Except for skin cancers, breast cancer is the most common malignancy among women, accounting for nearly one in three cancers diagnosed among women in the United States; it is the second leading cause of cancer death. Many countries in East Asia, including China also have a higher rate of breast cancer than before[1]. To date surgery, chemotherapy, radiotherapy, and hormone therapy play important roles in the treatment of breast cancer[2].Estrogen receptor-negative breast carcinomas do not respond to hormone therapy, making their effective treatment very difficult[3]. Recent studies reported studying peroxisome proliferators-activated receptor γ as a potential target for the prevention and treatment of human cancer. Artificial synthesis of PPARγ agonist thiazolidinedione (TZD) consisting of rosiglitazone, pioglitazone and ciglitazone has been widely used for the clinical therapy of diabetes mellitus[4]. Recent researches showed that TZD can restrain the growth of many tumor cells and induce tumor cell apoptosis, thus activating PPARγ. This may be a promising way in the treatment of many malignant tumors[5].
To the best of our knowledge, little is known about the anti-tumor effects of rosiglitazone on human breast cancer cells and the mechanism of its anti-tumor activities. In the present study, we have examined the growth inhibitory effects of rosiglitazone on human breast cancer cell line MDA-MB-231, and clearly demonstrated that rosiglitazone can inhibit MDA-MB-231 cell growth through the activation of PPARγ as well as cell cycle arrest and the induction of apoptosis in the high dose. These novel findings shed new light on its potential benefit in the treatment of breast cancer, especially in estrogen receptor-negative breast carcinomas.
Materials and Methods
Chemicals and reagents
Rosiglitazone, PPARγ selective antagonist GW9662, HEPES, dimethyl sulfoxid (DMSO), 3-(4,5-dimethylthiazolyl)-2,5-diphenyl tetrazolium bromide (MTT), trypsin, and propidium iodide (PI) were purchased from Sigma (USA). RPMI 1640, fetal bovine serum (FBS), penicillin and streptomycin were purchased from GIBCO (USA). Annexin V-FITC apoptosis detection kit and TUNEL assay kit are products from Beckman Coulter (USA) and Roche (Germany), respectively. Because rosiglitazone and GW9662 are difficult to dissolve, 0.1mol/L mother liquor was prepared with DMSO (stored at -20°C) and diluted with cell culture medium according to requested final concentration when it was used. The final DMSO concentration was always 0.1% in culture media.
Cells and cell culture
Human breast cancer cell line MDA-MB-231 was purchased from ATCC, USA. Cells were maintained in RPMI-1640 with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Cytotoxicity assay
The cytotoxic effect of rosiglitazone on MDA-MB-231 cells was measured by the MTT colorimetric method. Briefly, 1.2 × 104 cells were seeded in a 96-well plate for 15 h. Cells were exposed to various concentrations of rosiglitazone for different periods of time. Control wells received 0.1% DMSO vehicle at a concentration equal to that of drug-treated cells. 10 μl of 5 mg/ml of MTT was then added into each well and further incubated at 37°C for 4 h. After the medium and MTT were removed, 100 μl DMSO was added into each well and the absorbance was determined at 570 nm (reference wavelength at 630 nm) using a SAFIRE fluorescence microplate system from Tecan (Salzburg, Austria). The growth inhibiting rates of MDA-MB-231 cells were calculated with the following formula: Cell inhibitory rate = [1 - test group A570 nm/control A570nm] ×100%. 50% inhibiting concentration (IC50) was also calculated using IC50 calculation software. The morphologic features in MDA-MB-231 cells were observed using a microscope from Leica (Wetzlar, Germany).
Cell growth measurements
MDA-MB-231 cells were plated at a density of 5×104 per well in 6 well plates and replaced the next day with RPMI-1640 media containing 10% FBS and either 0.1% DMSO or the indicated drugs i.e. 1 μmol/L rosiglitazone, 5 μmol/L GW9662, or both rosiglitazone and GW9662 in the above concentrations. Cells were exposed to fresh media and media containing compounds every 48 h. The number of cells was counted at the 2, 4, 6 days posttreatment using a Coulter counter.
Cell cycle analysis
Using standard DNA labeling methodology, the DNA of cells treated with rosiglitazone was stained with PI, and the proportion of cells in various phases of the cell cycle was monitored by flow cytometer. Briefly, 3 × 105 cells were seeded in a 6-well culture plate. On the following day, the old medium was exchanged with fresh medium containing different concentrations of rosiglitazone (10-4, 10-5, 10-6 mol/L) and incubated continuously for 24 h. Cells were harvested by trypsinization and washed twice with cold phosphate-buffered saline (PBS). Cell pellets were fixed in 75% ice cold ethanol and stored at -20°C for at least 12 h. Fixed cells were centrifuged at 1000 rpm for 5 min, washed with cold PBS, and then stained with PI solution consisting of 45 μg/ml PI, 10 μg/ml RNase A and 0.1% Triton X-100 for 30 min to 1 h in the dark. The DNA content of stained cells was analyzed using CellQuest and ModFitLT 2.0EP Software with a FACSCalibur flow cytometry system from BD (USA).
Apoptotic assay
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay was used to further validate MDA-MB-231 cell apoptosis. The inoculated density of cells and drug concentrations used were the same as for cell cycle analysis. The TUNEL kit was used according to the manufacturer’s protocols. Cells were untreated or treated with rosiglitazone for 36 h prior to TUNEL staining assay.
Statistical analysis
All experiments were conducted in triplicate and all data were expressed as mean ± SD. The statistical differences were assessed by ANOVA analysis with the SPSS 13.0 software. Multiple comparisons among means were done with LSD t test. P values less than 0.05 were considered statistically significant.
Results
Cytotoxic activity of rosiglitazone on MDA-MB-231 cells
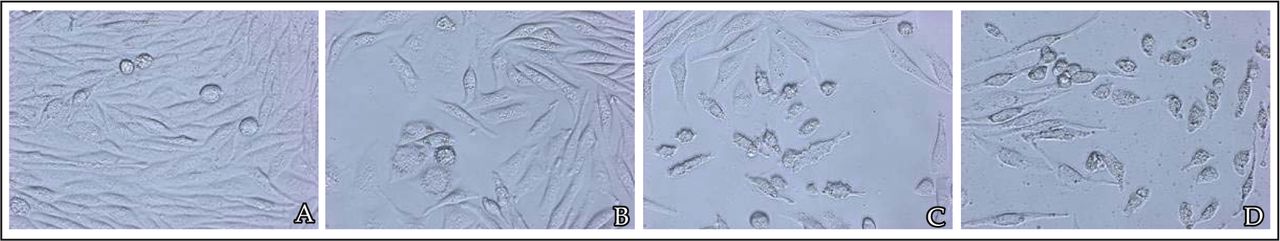
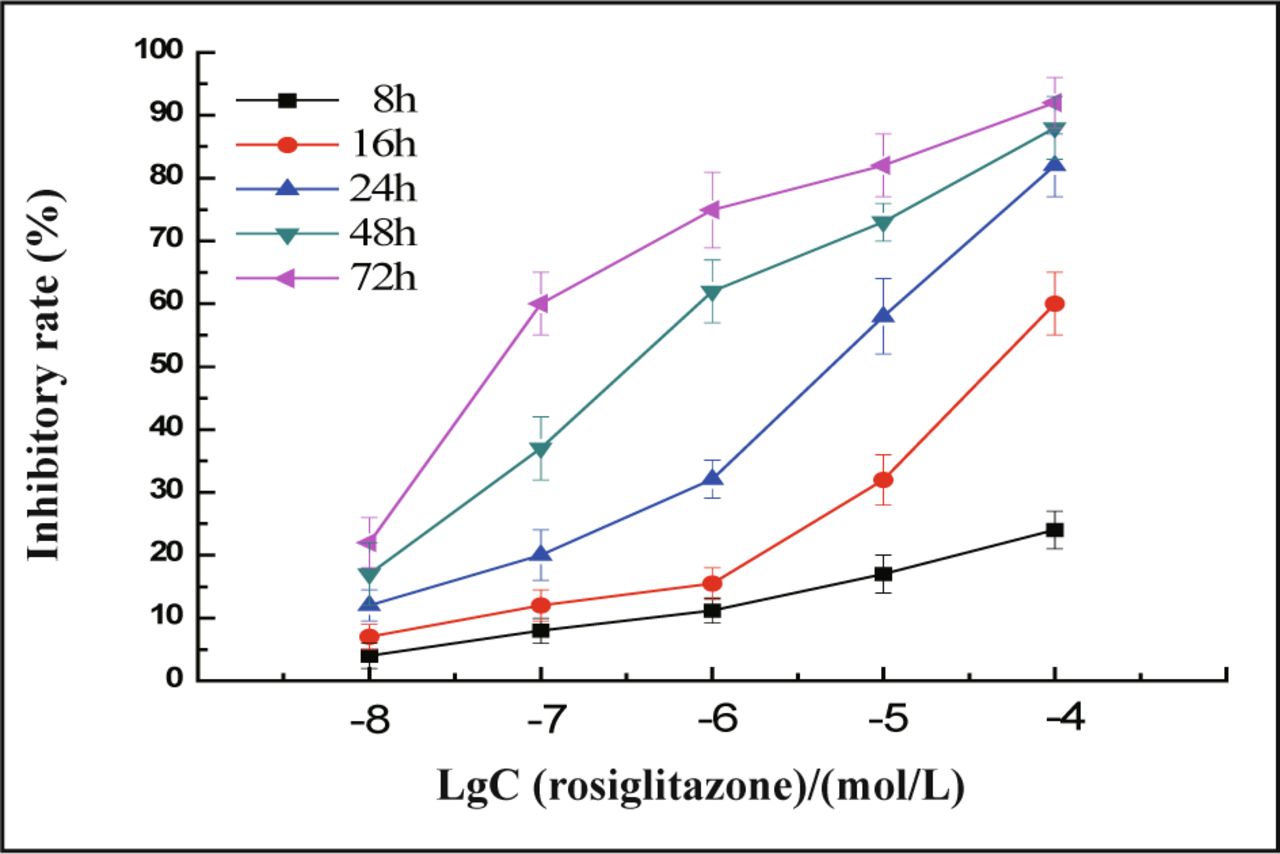
In the presence of different doses of rosiglitazone, the cells were inhibited to a greater extent ranging from 20% to 80% with a loss of viable cells (Fig.1). Rosiglitazone dramatically inhibited the proliferation of MDA-MB-231 cells in a concentration and time-dependent manner (Fig.2). The concentration which causes 50% inhibition of cell viability (IC50) by rosiglitazone was about 5.2 μmol/L at 24 h post-treatment.
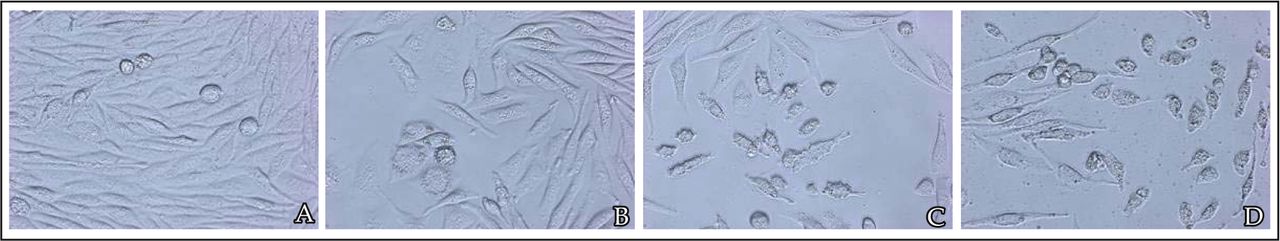
A, cells with 0.1% DMSO (untreated control); B, 10-6 mol/L rosiglitazone; C, 10-5 mol/L rosiglitazone; D, 10-4 mol/L rosiglitazone.
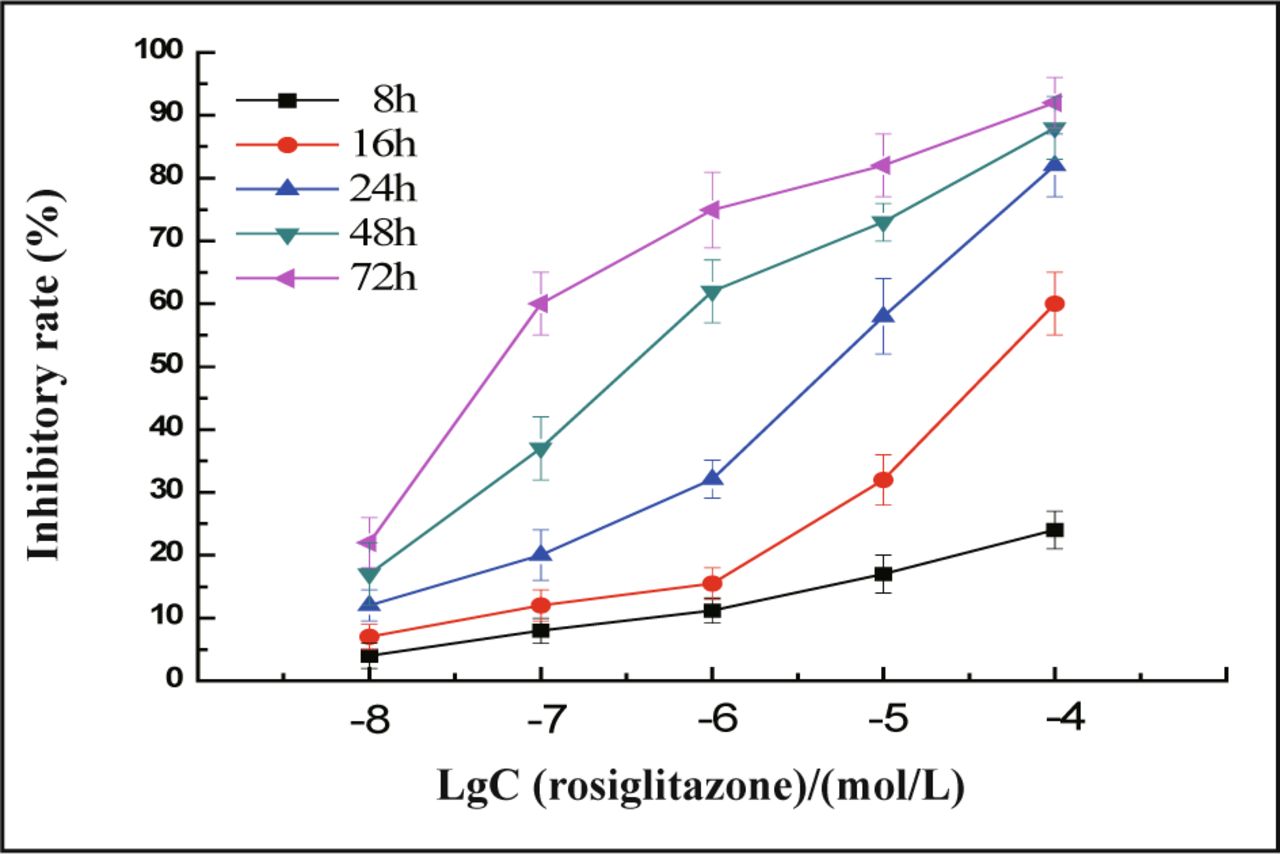
MDA-MB-231 cells were treated with different concentrations of rosiglitazone as indicated for 8 to 72 h, the inhibitory rate of cells was measured by the MTT. Rosiglitazone dramatically inhibits the proliferation of MDA-MB-231 cells in a concentration and timedependent manner. Values are mean and SD of triple determimations.
Anti-proliferation activity of rosiglitazone in MDA-MB-231 cells and its relationship to PPARγ
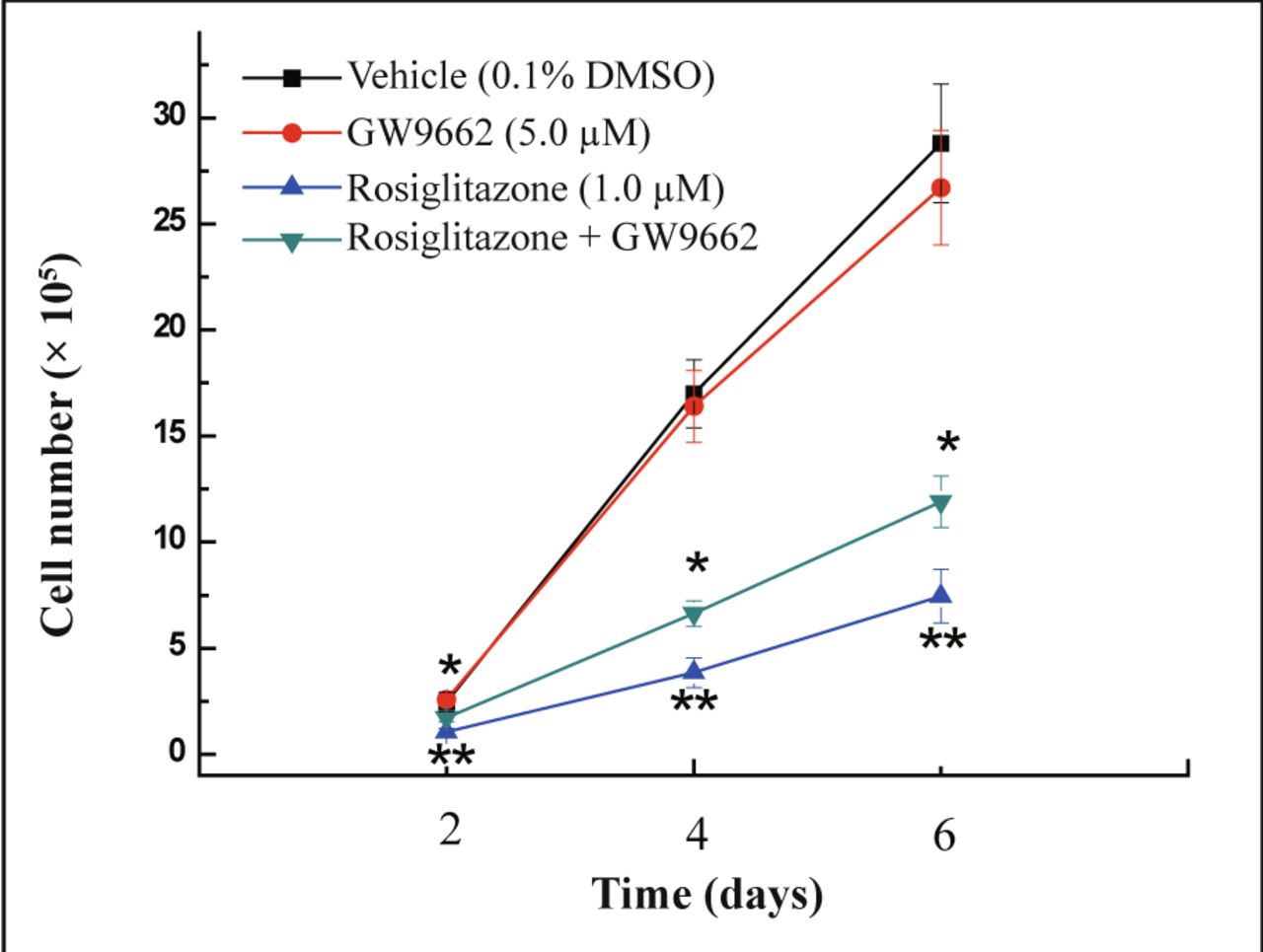
Cells were co-treated with rosiglitazone and PPARγ antagonist GW9662 to examine whether the growth inhibitory effect of rosiglitazone on MDA-MB-231 cells is dependent on activation of PPARγ. PPARγ agonist rosiglitazone induced a decrease in cell number compared with vehicle-treated cells and this decrease in cell growth could be reversed by co-treatment with GW9662 (Fig.3) (P < 0.05). Thus, the anti-proliferation activity of rosiglitazone in MDA-MB-231 cells was realized through activation of PPARγ activity.
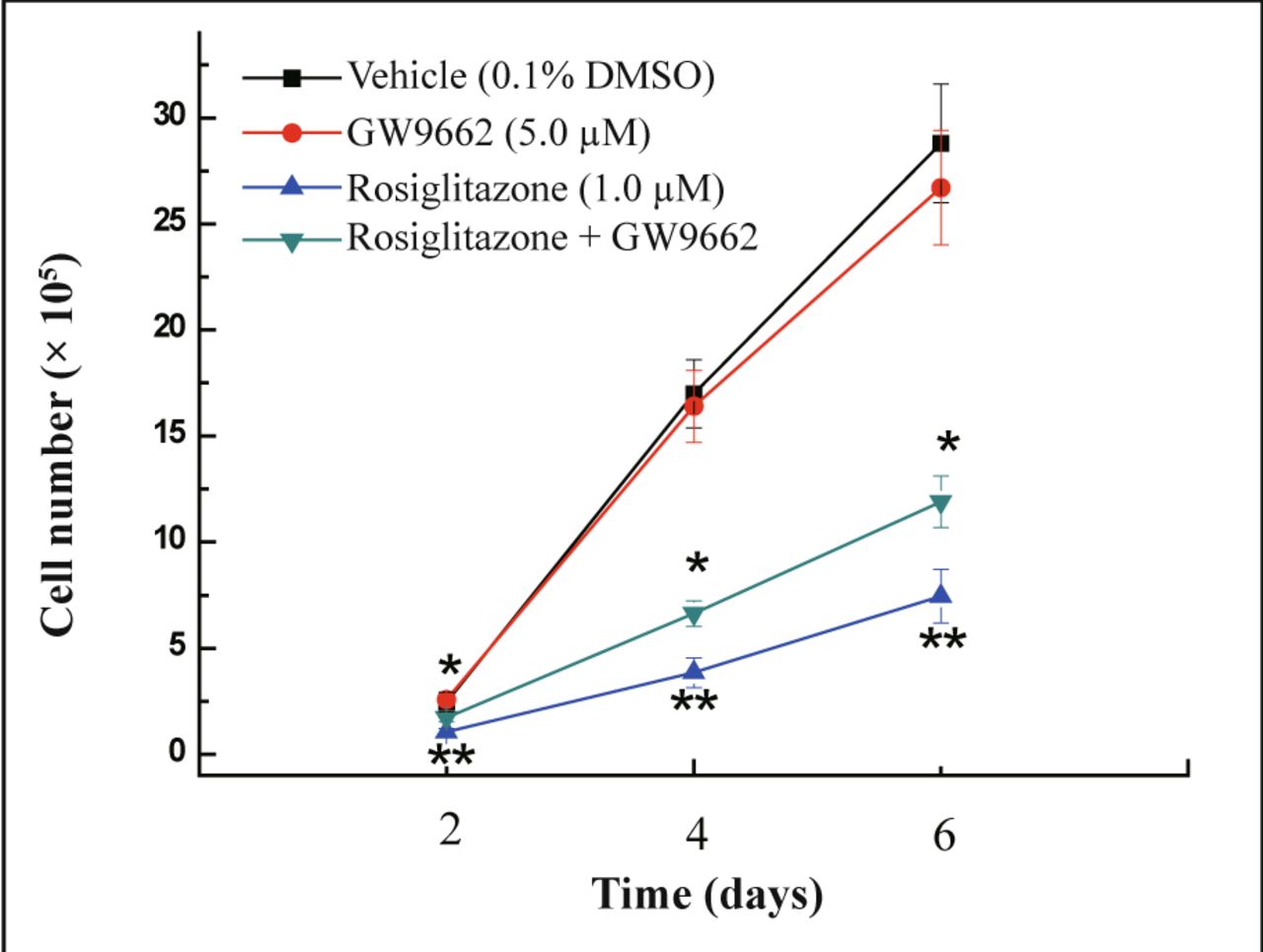
MDA-MB-231 cells were treated with the PPARγ agonists rosiglitazone (1.0 μmol/L), the PPARγ antagonist GW9662 (5.0 μmol/L), or in combination of rosiglitazone and GW9662. The number of cells was counted at 2, 4, and 6 days post-treatment. Each data point represents the mean of three independent experiments. *P < 0.05 as compared with the rosiglitazone group; **P < 0.01 as compared with 0.1% DMSO control group.
Cell cycle analysis
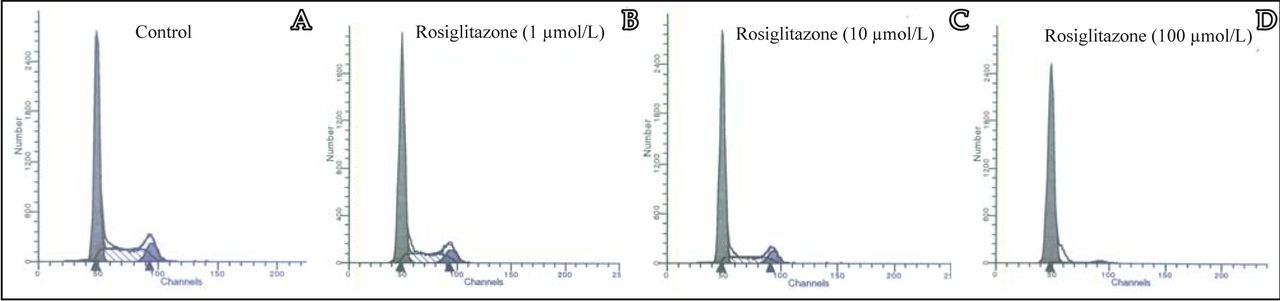
To examine whether the cytotoxicity of rosiglitazone was due to the induction of apoptosis by cell cycle arrest, cell cycle analysis was performed by FACSCalibur flow cytometry. After treatment of MDA-MB-231 cells with rosiglitazone for 24 h, the ratio of cells in the G0/G1 phase tended to increase, The number of cells in the S and G2/M phases decreased remarkably along with the increase of rosiglitazone as compared to the untreated control (Fig.4). These results suggested that rosiglitazone can induce cell cycle arrest at the G0/G1 phase.

MDA-MB-231 cells were treated with different concentrations of rosiglitazone as indicated for 24 h prior to flow cytometry analysis. A, Control group with 0.1% DMSO; B, 1 μmol/L rosiglitazone, C, 10 μmol/L rosiglitazone; D, 100 μmol/L rosiglitazone.
Apoptosis of MDA-MB-231 cells induced by rosiglitazone
To examine whether rosiglitazone inhibition of the proliferation of MDA-MB-231 cells was also partially due to the induction of cell apoptosis, TUNEL stain assay was used. TUNEL (Fig.5) assay staining gives apoptosis cells a brownish yellow nucleus. Apoptosis cells were found in the 10-4 mol/L rosiglitazone group and the apoptotic rate was (18 ± 3)%.
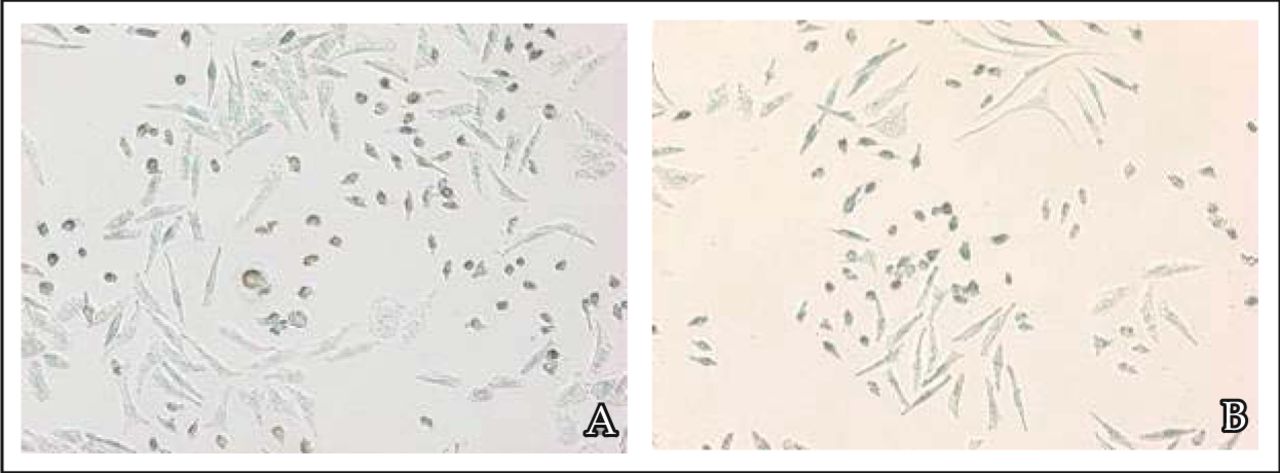
MDA-MB-231 cells were treated with 10-4 mol/L rosiglitazone for 36 h. A, Scattered apoptosis cells with nuclei stained by using the TUNEL staining assay. B, Negative control without using TdT enzyme during TUNEL staining.
Discussion
PPARγ is a member of the nuclear hormone receptor superfamily. It plays a critical role in a variety of biological processes, including regulation of metabolism, inflammation, cellular growth and differentiation[6]. Ligand-activated PPARγ acts as a transcription factor; forming a heterodimer complex with the retinoid X receptor (RXR), which then binds to the peroxisome proliferatior response element (PPRE) within the promoter of target genes and modulates their expression[7]. Scientists have paid much attention to the study of PPARγ in the past decade because of its important function in cell metabolic regulation[8.9]. Activating of PPARγ can increase insulin sensitivity, ameliorating abnormally high insulin resistance in patients with type 2 diabetes. The development and application of synthesized thiazolidinedione (TZD) PPARγ agonists include rosiglitazone, pioglitazone and ciglitazone which are currently promoted[4,10].
In recent years, PPARγ agonists including TZD have been reported to suppress the growth of cancer cells of diverse tissue origin[11-13]. Although the precise mechanism of PPARγ ligand-induced growth inhibition is not yet known, it has been found to be associated with induction of cell cycle arrest, differentiation of tumor cells and apoptosis as well as non-apoptotic cell death. In the study of human hepatoma cells, troglitazone could induce p21 expression, which is an inhibition factor of cyclin dependent protein kinase (CDK) and the hepatoma cells were arrested in G1 phase[14]. Studies on rat glioma cells showed that PPARγ agonists ciglitazone and PG-J2 could induce cell apoptosis by up-regulation of the apoptosis promoting proteins Bax and Bad and releasing cytochrome c[15].
Several lines of evidence suggested that breast tissues in both human and mouse as well as human breast cancer cell lines have PPARγ expression. In the present study, we further confirmed that human breast cancer cell line MDA-MB-231 has PPARγ expression. Yin and colleagues[16] found that PPARγ ligand troglitazone could inhibit MCF-7 cell proliferation by arresting cells in G1 phase. The accumulation of cells in G1 was accompanied by an attenuation of Rb protein phosphorylation associated with decreased CDK4 and CDK2 activities[16]. The present study shows that rosiglitazone inhibited MDA-MB-231 cell growth in a dose and time-dependent manner, 1 μmol/L rosiglitazone could significantly inhibit proliferation of MDA-MB-231 cells. Cell cycle analysis showed that rosiglitazone can inhibit the proliferation of MDA-MB-231 cells by G1 phase arresting. So we propose that the underlying mechanism of proglitazone on MDA-MB-231 cell proliferation is similar to the MCF-7 mentioned above.
MDA-MB-231 cells belong to estrogen receptor-negative breast cells which are insensitive to hormone therapy. Finding a new way to deal with this is, therefore especially urgent. So far, the studies involving TZD on MDA-MB-231 cells have been very limited. The phase II study which used troglitazone to treat breast cancer was stopped because of frequent incidence of hepatotoxicity[17]. Rosiglitazone is a high-affinity PPARγ ligand. Leung and colleagues found that rosiglitazone could inhibit MKN45 human gastric cancer cell growth in the nude mouse[18]. In anaplastic thyroid cancer, rosiglitazone-induced growth inhibition was associated with cell cycle arrest and changes in cell cycle regulators, such as an increase of cyclin-dependent kinases inhibitors p21 and p27, a decrease of cyclin D1, and inactivation of Rb protein. The rosiglitazone-induced apoptosis was associated with a decrease of Bcl-XL expression and caspase 3 and 7 activation[19]. A phase I study of rosiglitazone in patients with refractory cancers showed that human have a better tolerance to rosiglitazone and they did not detect any toxic effects[20]. In the 2005 annual meeting of American Cancer Association, Mody and colleagues reported that rosiglitazone could inhibit the proliferation of MDA-MB-231 cells[21].
More recently, a pilot study showed that short-term treatment with rosiglitazone does not significantly alter breast tumor cell proliferation in early-stage breast cancer patients. The lack of effect may relate to the short treatment period as well as the use of a lower dose of 4 mg twice a day. Indeed, the increase in serum adiponectin and the increase in insulin sensitivity point to potential risk reduction benefits with longer-term treatment[22]. In the present study, we not only confirmed that rosiglitazone could inhibit the proliferation of MDA-MB-231 cells but also found, for the first time, that the dosage of 100 μmol/L rosiglitazone could induce apoptosis of MDA-MB-231 cells. These findings provide new experimental proof for the clinical application of rosiglitazone in breast cancer therapy.
In some tumor cell lines, there was no correlation between growth inhibition sensitivity induced by TZD and expression level of PPARγ protein[23], making some scientists think that the mechanism of the anti-tumor effect of PPARγ agonist was independent of PPARγ activation[24]. Therefore, in this study, we investigated whether the growth inhibitory effects of rosiglitazone is truly dependent on transcriptional activation of PPARγ. The association between the growth inhibitory effect and transcription activation potential of PPARγ in MDA-MB-231 cell line was examined by co-treatment with rosiglitazone and PPARγ antagonist GW9662. The results illustrated that the inhibitory effect of rosiglitazone on proliferation of the MDA-MB-231 cell line was truly dependent on PPARγ receptor activation. Therefore, we propose that, at least in the case of MDA-MB-231 cells, the mechanism by which rosiglitazone inhibites proliferation of cancer cells is related to activation of the PPARγ receptor.
In conclusion, this study clearly showed that rosiglitazone could inhibit MDA-MB-231 cell proliferation via PPARγ activation. A high dose of rosiglitazone also induced cell apoptosis. Rosiglitazone could be a potential candidate to deal with breast cancer due to its significant anti-proliferative and apoptotic effects on MDA-MB-231 cells.
Acknowledgements
We are grateful to Xin Li for assistance in flow cytometry. We thank Dr. Jie Liu (Inorganic Carcinogenesis, NCI at NIEHS) for critical reading of this manuscript.
- Received June 13, 2008.
- Accepted October 10, 2008.
- Copyright © 2008 by Tianjin Medical University Cancer Institute & Hospital and Springer
References
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.