

Abstract
The genome of cells is constantly challenged by DNA damages from endogenous metabolism and environmental agents. These damages could potentially lead to genomic instability and thus to tumorigenesis. To cope with the threats, cells have evolved an intricate network, namely DNA damage response (DDR) system that senses and deals with the lesions of DNA. Although the DDR operates by relatively uniform principles, different tissues give rise to distinct types of DNA damages combined with high diversity of microenvironments across tissues. In this review, we discuss recent findings on specific DNA damage among different tissues as well as the main DNA repair way in corresponding microenvironments, highlighting tissue specificity of DDR and tumorigenesis. We hope the current review will provide further insights into molecular process of tumorigenesis and generate new strategies for cancer treatment.
keywords
Introduction
DNA damage is a serious threat to human health. It has been estimated that each cell in human body experiences approximately 100,000 DNA damage events per day1. These damages include occasional DNA mismatches during DNA replication in physiological processes, DNA strand breaks caused by aberrant topoisomerase (Topo) I and Topo II activity, and DNA lesions caused by reactive oxygen species (ROS)2,3. In addition, various stressors from the surrounding environment, such as ionizing radiation (IR) and ultraviolet (UV) light, as well as chemical factors including heavy metals, alkylating agents, nucleoside analogs and base analogs, can attack DNA, resulting in adduct formation, base deletion, DNA strand breakage, or crosslinking4-6. Some chemical agents are highly electrophilic and can react directly with DNA, while others must be metabolized before they can insult the nucleic acids7. DNA lesions can block DNA replication and transcription8. If not repaired timely and accurately, they may cause genomic instability or chromosomal abnormalities that threaten the integrity and functions of cells, tissues and organs9-11.
Living cells must constantly address genotoxic insults through a complex and coordinated process, known as DNA damage response (DDR) system12. The highly conserved DDR network consists of hierarchical pathways. The crucial components of the network can generally be classified as damage sensors, transducers, and effectors2. The DDR network can actively sense and signal problems in DNA to induce different cellular effects13. The fate of damaged cells is greatly influenced by multiple factors such as the extent of DNA lesions, the choice of DDR pathways, the rapidity and fidelity of DNA repair, and the status of the cells. Depending on these conditions, cells may temporarily fall into cell cycle arrest for DNA repair, or irreversibly undergo senescence or apoptosis6. Even failing to repair DNA lesions does not necessarily lead to fixation of potentially detrimental mutations, as such genetically aberrant cells are generally eliminated from the proliferative pool. This means that, DDR serves as a guardian of genomic integrity in a fairly broad sense.
While the association between defective DDR and predisposition to carcinoma has been recognized for decades, recent studies have revealed a novel role of DDR as a physiological barrier against tumorigenesis and tumor progression from early stages to advanced, invasive lesions14,15. Tumorigenesis is a multi-step process. An early step is the dysregulation of cell proliferation resulted from oncogenic activation or loss of tumor suppressors, which subsequently triggers DDR16. Such DDR activation achieves urgent disposal of precancerous cells, posing the cells for repair, senescence, or cell death. Thus, the process helps to delay or avoid uncontrolled cell growth and tumorigenesis. When progressing from pre-neoplastic abnormalities to cancers, this barrier has been observed to be diminished in most tissues through loss of certain DDR capabilities17. On the other hand, a cell with DDR deficiency indeed displays higher genomic instability and increased dependency on remaining DDR pathways, leading to a worse outcome during its DNA repair. This vicious circle overloads threats on the genome of the cell that may initiate its tumorigenesis.
General DNA damage and repair
In mammalian cells, major DNA repair types include nucleotide excision repair (NER), base excision repair (BER), mismatch repair (MMR), double-strand break (DSB) repair through homologous recombination (HR) or non-homologous end-joining (NHEJ), and Fanconi anemia (FA) DNA repair18. Single-strand breaks (SSBs) in which a 3'-hydroxyl adjoins a 5'-phosphate without missing nucleotides can be directly ligated19. Before discussing the features of DDR in different tissues, we briefly introduce the general functions of these DNA repair ways.
The NER pathway removes bulky adducts induced by UV light or platinum salts, and also the modified nucleotides that distort the structure of the double helix20,21. The two subpathways of NER, global genomic NER (GG-NER) and transcription-coupled NER (TC-NER), start differently22. The former is initiated by XPC and DDB1-DDB2 complex, while the latter relies on RNA polymerase II, CSA, CSB, and TFIIS to sense lesions23-25. Both of the subpathways lead to the recruitment of transcription factor II human (TFIIH) to unwind the DNA helix at the damaged site26. Xeroderma pigmentosum group A (XPA) and replication protein A (RPA) then orchestrate this open complex formation and stabilize the repair intermediate. The xeroderma pigmentosum group F (XPF) protein and the excision repair cross-complementation group 1 (ERCC1) protein form a designated complex, XPF-ERCC1, to cleave 5' of the lesion, while xeroderma pigmentosum group G (XPG) executes the 3' incision27. Replication factor C (RFC) then loads proliferating cell nuclear antigen (PCNA) to accommodate DNA polymerases for repair replication. And the final ligation step is carried out either by flap endonuclease 1 (FEN1) and DNA ligase I (LIG1) or by the ligase III-XRCC1 complex28. Different from NER, the BER pathway deals with small chemical alterations of DNA bases, abasic sites, and SSBs29. BER is initiated by one of at least 11 different damage-specific DNA glycosylases with or without apurinic/apyrimidinic endonuclease 1 (APE1)30. Glycosylases cleave the base-sugar bond, leaving an abasic site31. The resulting SSB can be filled in and ligated via short-patch or long-patch BER. The former recruits X-ray repair cross-complementing protein 1 (XRCC1), DNA ligase III (LIG3), poly ADP-ribose polymerase 1 (PARP-1), and DNA polymerase ݠ(pol ݩ to fill the gap with a single nucleotide32. The latter loads polymerases that synthesize at least two nucleotides for long-patch gap filling, and this process requires PCNA and RFC33. Finally, the displaced strand is excised by FEN1, and the incision is ligated by LIG134. NER and BER are both carried out by multiprotein complexes comprised of approximately 20 components. Besides, both of the pathways require incision of the damaged DNA strand, with subsequent re-synthesis using the complementary strand as a template35. The size of repair patch differs, however, in that the NER patches are approximately 30 bases in length, whereas BER produces the patches of 1-6 bases in length36. Remarkably, NER and BER are carefully regulated by p53. p53 affects NER via the transcriptional activation of downstream effector genes including p48-XPE and Gadd45a37,38. As for BER, p53 might directly interact with BER proteins, including pol ݠand APE39-42. These effects are believed to be fundamental for p53 tumor suppressor to repair the vast majority of DNA damages incurred from various environmental stressors43.
MMR is another important DDR pathway for resisting tumorigenesis. This pathway mainly corrects replication errors, including nucleotide substitutions, deletions, and insertions. MMR is initiated when a heterodimer of the MutS homolog (MSH), MSH2-MSH6 (MutSa), recognizes a base-base mismatch or when MSH2-MSH3 (MutSݩ recognizes an insertion-deletion loop (IDL) longer than two base pairs44,45. These heterodimers recruit the postmeiotic segregation 2 (PMS2) protein bound to either MutL homolog 1 (MLH1) or MutL homolog 3 (MLH3), and the interactions activate PMS2 endonuclease activity46. Exonuclease 1 (EXO1) excises the DNA segment containing the mismatch, while RPA coats the single-stranded DNA (ssDNA). DNA polymerase works to fill the gap, and the DNA is then ligated. MMR pathway is usually robust in dividing cells, as mismatches occur frequently at the replication fork47. In accordance with this understanding, the genomes of MMR-deficient carcinomas are revealed to harbor considerable amount of somatic mutations48. Based on the indication that these MMR-deficient cancers may produce mutation-associated neoantigens recognized by the immune system, novel studies on immune checkpoint therapy were carried out, and further suggested that advanced solid tumors with MMR deficiency were sensitive to programmed death receptor-1 (PD-1) blockade48-50.
Among different types of DNA damage, DSBs are the most deleterious DNA lesions that may result in genome rearrangements, chromosomal translocations, and cell death. In mammalian cells, DSBs are repaired through either NHEJ or the HR process51,52. NHEJ, rapid but error-prone, is the main DSB repair option that may occur in each stage of the cell cycle53. The initiator of NHEJ, Ku70/80 heterodimer, has high affinity for the ends of DSBs54. DNA-bound Ku recruits DNA-protein kinase catalytic subunits (DNA-PKcs) to DSB ends to form a DNA-PK complex, activating the kinase activity of DNA-PKcs and facilitating ligation55. If the DSB ends are not compatible for ligation, end-processing enzymes are necessary for resecting the DNA ends, filling the gaps, and removing the blocking end groups. Crucial enzymes in this process include polynucleotide kinase 3'-phosphatase (PNKP), aprataxin and PNK-like factor (APLF), Artemis, polymerases µ and γ, and Werner (WRN)56. Finally, DNA ligase IV complex, consisting of DNA ligase IV (LIG4), X-ray repair cross-complementing protein 4 (XRCC4), and XRCC4-like factor (XLF), is responsible for ligation57. Despite its protective role in DSB lesions, NHEJ has been proposed to be involved in the formation of chromosomal translocations and act as a key source of genomic rearrangements and instability, especially when HR or other DDR pathways are defective58,59. Cells with mutations of genes in error-free repair pathways, such as XRCC2, FANCC, and BLM(RECQL3), may overload NHEJ for repair, resulting in a higher risk of cancer60-62.
Compared to NHEJ, HR-mediated DSB repair is more accurate. However, HR relies on the presence of sister chromatids, limiting the use of this pathway to cells in late G2 or S phase. In HR, DSBs are detected by Mre11-Rad50-Nbs1 (MRN) complex, which activates the ataxia telangiectasia mutated (ATM) checkpoint kinase for efficient phosphorylation of DNA repair factors. As MRN triggers 5' to 3' end resection of DSB ends, the range of resected DNA is extended, giving rise to single-stranded 3' overhangs63,64. The ssDNA ends are rapidly bound and protected by RPA and are subsequently replaced by Rad51 recombinase for the generation of long nucleoprotein filaments65. Using the DNA strand of the undamaged chromatid as a template, Rad51 filaments assist in the homology search. After recombination repair, the DNA ends are joined by ligases. On the basis of HR mechanism, numerous new proteins and pathways with functions in HR have been identified in the past decades. For example, accumulated DNA damage due to inactivation or loss of BER, NER or translesion synthesis might be resolved or bypassed by HR during replication66-68. It is currently a consensus that overall DDR network, cell cycle checkpoints, and chromatin remodeling are critical for the onset, regulation, and efficiency of HR69-71. Due to its potent DSB execution, high repair fidelity, and broad crosstalk among pathways, HR can be viewed as the most important repair mechanism and a last resort for DNA repair. Tumors with mutated HR genes are usually characterized by gross gene rearrangements72.
Genes mutated in FA patients are found to interact with the DNA repair genes BRCA1 and FANCD1 (BRCA2) upon replication stalling and thus suppress tumorigenesis73. In particular, the FANCM-FAAP24-MHF1-MHF2 anchor complex detects lesions caused by interstrand crosslinks (ICLs), and recruits the core complex when activated74. The FA core complex is composed of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and the accessory proteins FAAP20 and FAAP10075. The complex monoubiquitylates each protein of the FANCI-FANCD2 (ID2) heterodimer. This process leads to the formation and loading of the ID2 complex at nuclear foci76,77. Ubiquitylated ID2 facilitates the DNA breakage at ICLs and translesion synthesis, and then signals downstream HR proteins such as FANCD1 (BRCA2), FANCJ (BRIP), FANCN (PALB2), and FANCO (RAD51C) to repair the collapsed replication fork induced by strand break78-83. Besides, FANCD2 mediates fork protection via RAD51 functions, which indicates a novel pathway connecting FA components to RAD51 and the BRCA1/2 tumor suppressors84. However, FA proteins are not classic HR factors, and cells from FA patients do not demonstrate severely defective HR repair of DSBs85. Therefore, the functional relationship between FA and HR proteins during replication stalling remains ambiguous and needs further exploration.
Tissue-specific DDR and tumorigenesis
Because of their surrounding environments, organs or tissues are faced with different insults which lead to various types of DNA damage86. Some tissues, such as the small intestine, are particularly sensitive to DNA damage, yet the colon is considerably more resistant87,88. While different pathways cooperate to accomplish DDR, cells with distinct origins differ in their capacities to sense, respond to, and resolve DNA lesions. For example, rapidly proliferating cells of the small intestine and colon have an efficient MMR pathway, while the NER, NHEJ, and HR pathways are more frequently initiated in skin cells for the repair of ICLs caused by UV radiation89-91. Clinical and experimental findings have provided extensive evidence for tissue-specific DNA repair. Mutations of DNA repair genes in different DDR pathways tend to have tissue-specific effects on patients, leading to tissue-specific phenotypes92-94. For example, variations in MMR pathway, particularly when involving MSH1, MSH2, MLH6, or PMS2, tend to increase the risk of colorectal cancers (CRCs), whereas mutations in SSB repair mechanism can have a higher chance to impair neurological functions94,95. People with mutations of XPA, XPC, XPE, and XPF in NER predispose xeroderma pigmentosum (XP) and subsequently cutaneous neoplasms, while hereditary or sporadic BRCA1, BRCA2, PALB2 and other HR mutations indicate greater incidence of breast and ovarian cancers93,96. It seems that these differences arise in accordance with the distinct characteristics of tissue cells. MMR plays an essential role in dividing cells of gut because of frequent mismatches at DNA replication fork. By contrast, neurons are more sensitive to SSBs and oxidative damage due to their postmitotic status and high metabolic rates, thus more dependent on corresponding repair. Sequencing of cancer genomes has in turn revealed a wide range of mutational signatures that vary in part due to their tissues of origin97.
Furthermore, transgenic mouse models with reporter genes such as lacZ and lacI were used to quantify mutagenesis, and the results of the study have confirmed that mutation frequency varies considerably in a tissue-dependent manner98. Accordingly, the steady-state amount of oxidized DNA bases is proved to be higher in brain than in liver tissues, particularly in older mice99. By using oligonucleotide substrates to harbor SSBs in the nucleus and mitochondria, scientists have even found that testes contain higher glycosylase activity for BER than other tissues100. Though the precise underlying mechanism of tissue specificity in DDR and DNA repair has not yet been fully elucidated, many advanced findings have emerged and impelled our understanding in this field. Here, we will discuss the typical causes and types of DNA damage, the corresponding DDR or other DNA repair mechanisms, and the tumorigenesis in different tissues, hoping to provide further insights into DNA damage-induced tumors and new strategies of cancer treatments based on tissue specificity.
Skin
Skin is the largest organ of the human body, as it constitutes approximately 16% of the body mass101. Covering the body surface and serving as a protective barrier to isolate the internal environment of the body from the external environment, it plays a crucial role in resistance to daily stresses, such as harmful UV radiation from the sun, chemical agents, and infectious pathogens102. Remarkably, as many as 105 UV-induced photolesions caused by sunlight exposure are estimated to occur per day in every skin cell103. Ambient sunlight mainly contains UV-B (295320 nm) and UV-A (320400 nm), and each component can cause DNA damage through different mechanisms104. By interfering with nucleotide base pairing, UV radiation contributes to direct DNA photolesions, particularly (6,4)-photoproducts and cyclobutane pyrimidine dimers in skin cells105. These types of damage are mainly resolved through the NER pathway, indicating the significance of NER in skin disorder resistance106.
A large variety of skin maladies could be aroused if NER fails to repair DNA lesions in a timely manner107,108. The three most common diseases related to defective NER are XP, Cockayne syndrome (CS), and trichothiodystrophy (TTD). XP is a disease characterized by photosensitivity, ichthyosis (dry and scaly skin), and aberrant hyperpigmentation in skin areas exposed to sunlight. Notably, XP augments the susceptibility to UV-induced skin cancer (melanomas, basal and squamous cell carcinomas in particular) by 1000-fold109. Numerous mutations in genes, including XPA, ERCC3 (XPB), XPC, ERCC2 (XPD), DDB2 (XPE), ERCC4 (XPF), ERCC5 (XPG), and POLH (XPV), have been found in XP cells. These genes serve primarily to encode XP repair proteins in the NER pathway93,105. Depending on how much the mutations affect NER, different mutations can result in great heterogeneity in clinical phenotypes, and symptoms of XP can vary substantially in severity110. Although skin photosensitivity is presented in all three conditions, CS and TTD, unlike XP, have been found to be unrelated to increased skin cancer risk111. CS can be ascribed to mutations in ERCC8 (CSA), ERCC6 (CSB), XPB, XPD or XPG that induce defects in TC-NER. TTD is identical to CS, except for its additional cutaneous symptoms, including ichthyosis and brittle hair and nails. Mutations in TTDA, XPB, or XPD are the main genetic causes of TTD. Since these genes encode subunits of TFIIH, mutations in the genes give rise to destabilization or even dysfunction of the transcription factor112.
Cutaneous neoplasms are largely confined to patients with mutations in XPA, XPC, XPE and XPF93. Patients with CS due to defects in TC-NER, though photosensitive, are not prone to cancer, indicating the primary role of GG-NER in protecting DNA against UV-induced mutagenesis. Several studies on the effects of UV in NER-deficient mice have provided further evidence that Xpa-/-, Xpc-/- and Xpe-/- mice are prone to UV-induced skin cancer113-115. Moreover, Xpc-/- mice, defective merely in GG-NER, are not susceptible to edema or erythema, whereas Xpa-/- mice, which are defective in both GG-NER and TC-NER, are predisposed to both erythema and skin cancer induced by UV-B114,116. Thus, it can be concluded that, in GG-NER deficiency, mutations accumulate in areas of the skin exposed to UV radiation and prompt skin tumorigenesis117. By contrast, in most cases, TC-NER serves merely as an auxiliary mechanism against the cytotoxicity of DNA lesions118. Edema and erythema, rather than skin cancer, tend to be induced by UV when TC-NER is not intact. In addition, UV can cause mutations indirectly by generating ROS, such as hydrogen peroxide, hydroxyl radicals, and superoxide anions. ROS accumulation easily leads to oxidative lesions and SSBs119,120. The BER pathway is the main mechanism by which free radical lesions in DNA are repaired to prevent oxidative mutagenesis. Thus, defects in the BER pathway can also adversely affect skin121.
Liver
The liver is a highly specialized organ in vertebrates that mostly consists of hepatocytes122. Located in the right upper quadrant of the abdomen and inferior to the diaphragm in humans, the liver is believed to carry out as many as 500 different functions, typically in collaboration with other organs or systems123. This organ detoxifies various metabolites, synthesizes proteins, and produces bile, which aids in digestion by emulsifying lipids. The liver also plays a crucial role in metabolic processes, including the production of hormones, decomposition of red blood cells, and regulation of glycogen storage124-126.
Hepatic carcinoma is the sixth most prevalent cancer worldwide and the third leading cause of cancer death, of which hepatocellular carcinoma (HCC) is the principal histological subtype127. The development and progression of HCC is closely related to multiple risk factors, such as hepatitis B and C virus (HBV and HCV, respectively) infection, diabetes and obesity, aflatoxin exposure, alcohol consumption, diet contamination, and high levels of sex hormones128-130. These risk factors have been suggested to directly or indirectly foster the formation of DNA adducts and the production of ROS and/or reactive nitrogen species (RNS). For instance, hydroxyl radical has been shown to be the most damaging species that is responsible for numerous base modifications including thymine glycol, thymidine glycol, 5-(hydroxylmethyl)uracil, and 8-hydroxydeoxyguanosine131. In patients of chronic hepatitis C, higher level of 8-hydroxydeoxyguanosine in DNA extracted from liver tissue has been found132,133. These risk factors can potentially give rise to genome instability and thereby lead to neoplastic transformation. They may also target certain genes of DDR and/or DNA repair pathways. Aberrations in DNA repair proteins involved in HR, NHEJ, NER, BER, and MMR pathways, including p53, XRCC1, OGG1, ATM kinase, MRN complex, and PARP-1, have been reported to be associated with the development of HCC134-137.
Specifically, chronic HBV infection is one of the dominant risk factors for HCC development. It facilitates carcinogenesis via direct and/or indirect mechanisms. Direct effects of HBV include the interplay between HBV x protein (HBx) and host proteins. Viral DNA integration into the host genome can also arouse multitudinous mutations and chromosomal instability138. Indirect oncogenic effects of HBV involve oxidative stress and chronic inflammation. These events lead to accumulation of both genetic and epigenetic abnormalities in the liver, and result in chronic hepatitis, fibrosis, cirrhosis, tumorigenesis and tumor progression139. Chronic inflammation is characterized by the release of free radicals, the recruitment of immune cells to the liver, and the overexpression of cytokines such as interleukin 6 (IL-6), tumor necrosis factor a (TNF-a), and transforming growth factor ݠ(TGF-ݩ140,141. In addition, according to cohort studies, diabetes and obesity increase the risk of HBV-associated HCC142,143. Aflatoxin B1 (AFB1), an established potent hepatocarcinogen, is metabolized to an active intermediate that binds to DNA. This process fosters a typical AGG to AGT transversion at the 249th codon of p53, and thus boosts clonal expansion of mutant cells144,145. A better understanding of relevant DDR factors in HCC can help us develop new strategies for HCC prevention and treatment.
Pancreas
The pancreas is a vital organ composed of exocrine and endocrine parts, and the exocrine part comprises more than 80% of the pancreatic mass146. The exocrine pancreas is constructed from a branching network of acinar and duct cells. To aid in protein regulation and carbohydrate digestion, the exocrine pancreas secretes digestive zymogens into the duodenum, and produces high concentrations (higher than 140 mM) of bicarbonate (HCO3) in the pancreatic duct147,148. The endocrine components, on the other hand, include four types of specialized endocrine cells: a, ݬ d, and pancreatic polypeptide (PP) cells. These cells gather into clusters called islets of Langerhans. Through secretion of different hormones into the bloodstream, the endocrine pancreas precisely regulates metabolism and glucose homeostasis146.
Pancreatic cancer is one of the most malignant and aggressive human cancers worldwide149. The average survival of pancreatic cancer patients is barely 6 months, and the five-year mortality is 97%98%, commonly due to widespread metastasis150,151. In recent decades, studies on pancreatic cancer have detected certain types of DNA damage derived from exogenous carcinogen exposure and endogenous metabolic processes152. For example, aromatic DNA adducts and other types of DNA damage induced by smoking have been identified in the human pancreas153-155. Moreover, mutations in DDR factor genes, including BRCA2, PALB2, ATM, DNA-PK, CHK1, CHK2, FANCC, and FANCG, have been proven to be associated with chronic pancreatitis and pancreatic cancer149,156,157. In particular, mutations in BRCA2 and its partner PALB2 in HR are often involved in hereditary pancreatic cancer, indicating the key role of HR in suppressing hereditary pancreatic tumorigenesis158,159.
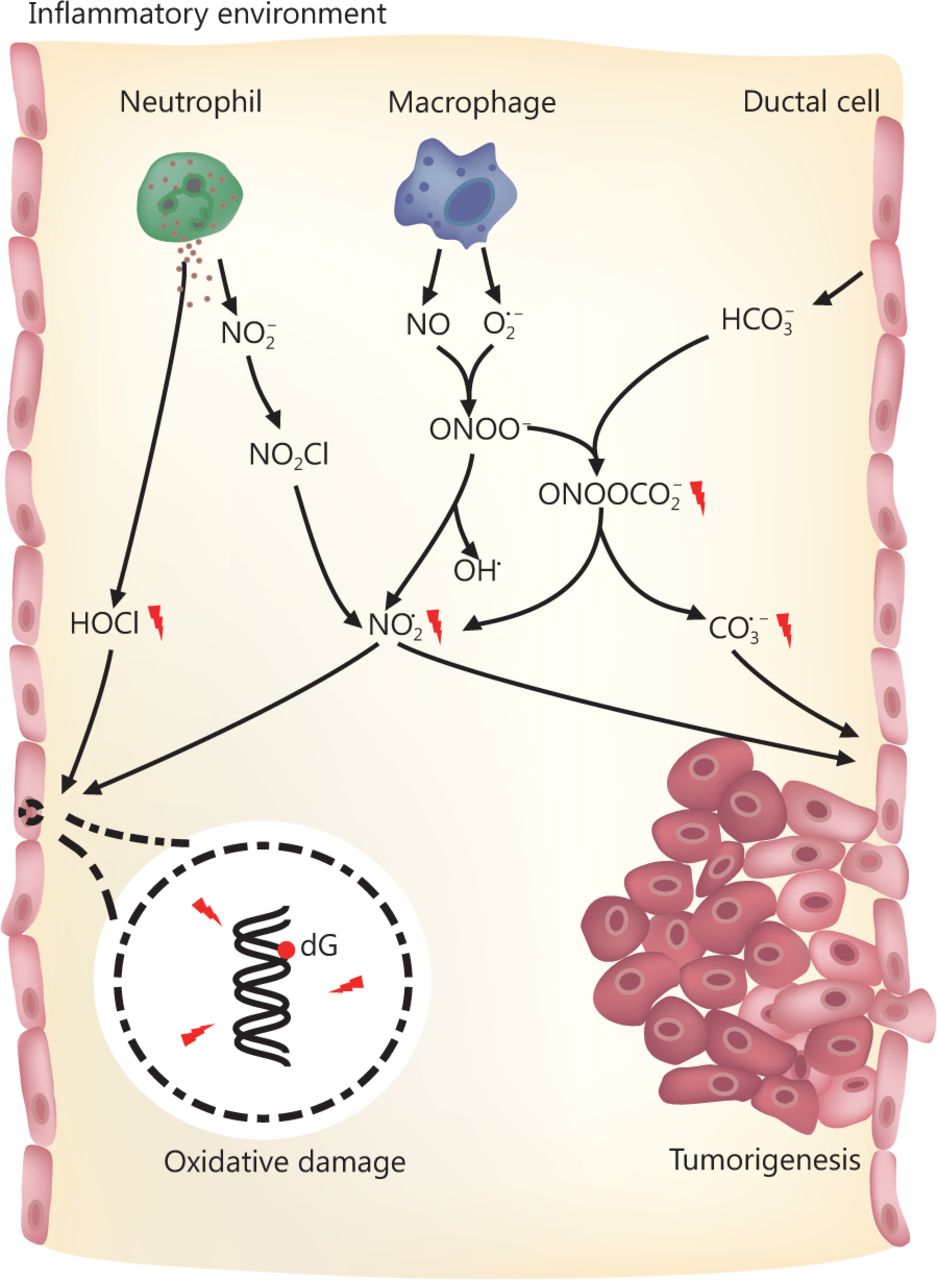
Established risk factors for pancreatic cancer include family history, aging, and smoking. Acute and chronic inflammation are also related to pancreatic cancer, especially hereditary pancreatic cancer160162. During the development of pancreatitis, neutrophils and macrophages are recruited and activated163. These cells can generate large quantities of ROS and RNS to eradicate pathogens164,165. Many of the ROS and RNS are carcinogens that can potentially arouse structural alterations in DNA, interfere with signal transduction pathways, or affect the activity of stress-response genes166168. For instance, the activity of myeloperoxidase (MPO) in neutrophils yields hypochlorous acid (HOCl), a potent oxidizing and halogenating agent169. In addition, MPO mediates the conversion of nitrite (NO2) into the nitrogen dioxide radical (NO2·) through a nitryl chloride (NO2Cl) intermediate170. Activated macrophages can secrete the unstable substances nitric oxide (NO) and superoxide (O2·), which then generate peroxynitrite (ONOO) through interaction171. Though not a free radical itself, ONOO homolyzes (t1/2 ~ 1 s) to form OH· and NO2·, thus causing 2-deoxyribose oxidation in DNA172. ONOO also reacts with high concentrations of HCO3 to produce nitrosoperoxycarbonate (ONOOCO2), a significant RNS in the pancreas. ONOOCO2 undergoes rapid homolysis (t1/2 ~ 50 ms) to generate the free radical NO2· and carbonate radicals (CO3·), which may pose strong threats to genomic integrity and stability173. Deoxyguanosine (dG) has the least reduction potential among the four types of DNA deoxynucleosides, the produced radicals thereby preferentially cause nitration and oxidation of dG in DNA and probably of guanosine (G) in RNA164,174 (Figure 1). Low levels of oxidative lesions in DNA are quickly repaired via the BER pathway in most tissues175. However, many more RNS are formed in concentrated HCO3 in pancreatic ducts once inflammation occurs158. Excessive damage may overload the BER pathway and induce DNA SSBs. Accumulated SSB lesions can be duplicated during DNA replication and thus be converted into DSBs. Alternatively, DSBs may also be produced during BER if two SSBs in complementary strands are located close to each other3. When tissues bear gene mutations that impair HR, such as BRCA2 and PALB2 mutations, they are highly threated by these DNA lesions and may develop cancers.

RNS and ROS in pancreas and the potential risk of tumorigenesis. When pancreatitis occurs, macrophages and neutrophils in pancreas secrete a large amount of nitric oxide (NO), superoxide (O2·) and nitrite (NO2) for the eradication of microbial pathogens and processing dead cells. These molecules are unstable and react with each other or microenvironment to produce hypochlorous acid (HOCl) and nitrosoperoxycarbonate (ONOOCO2), generating RNS and ROS including nitrogen dioxide radical (NO2·), and carbonate radical (CO3·). The reactive species have significant side effects on normal cells such as ductal cells in pancreas via attacking their genomic DNA. This poses great threat on genomic integrity and potentially causes pancreatic cancer.
Colon
The colon is the terminal part of the digestive system. Unlike the small intestine, which functions primarily to absorb nutrients, the colon extracts water and salt from unabsorbed materials before they are discharged from the body. The colon is also the site of flora-aided fermentation. At the microscopic level, the colon is lined with a simple columnar epithelium with invaginations called colonic crypts. It has been estimated that each colonic crypt contains 1,5004,900 cells, 56 of which are stem cells at the crypt base176. After cells are produced at the crypt base, they migrate upward along the axis of the crypt before detaching into the colonic lumen several days later177.
Due to its structural features and roles in the body, the colon is constantly challenged by tumorigenic factors, including dietary factors, gut microbiota-related factors, inflammation, and genetic mutations. As mismatches occur frequently at the replication fork in dividing cells, the MMR pathway is especially crucial in the colon47. Mutations in MMR genes typically lead to changes in the lengths of tandem nucleotide repeats located throughout the genome, producing a hypermutable phenotype known as microsatellite instability (MSI)178. MSI is found in approximately 15% of CRCs179. During replication, alterations in tandem nucleotide repeats give rise to temporary IDLs in DNA. If not corrected by MMR, these IDLs can generate frameshift or missense mutations in coding genes during subsequent rounds of DNA replication, ultimately resulting in the translation of aberrant proteins. In recent studies, MMR has also been indicated to be involved in multiple processes aside from DNA repair, such as miRNA processing and apoptosis induction180-182.
CRC is the third most prevalent cancer worldwide, leading to extensive morbidity and over 690,000 mortalities annually183. CRC progression is a gradual process in which normal colon epithelial cells (CECs) develop into polyps or colorectal adenoma and increasingly develop into colorectal adenocarcinoma184. Most CRC cases are sporadic, while approximately 30% of reported CRC cases are familial, with merely a small proportion having been well characterized185. Germline mutations in MMR genes, most commonly MLH1, MSH2, MSH6, and PMS2, have been proven to give rise to Lynch syndrome (also known as hereditary nonpolyposis colorectal cancer, HNPCC), the most prevalent form of hereditary CRC179. The majority of CRCs with high-frequency MSI (MSI-H), however, are sporadic non-Lynch syndrome cases186. These cases are caused by epigenetic inactivation of the MLH1 promoter via DNA hypermethylation. This inactivation often arises in tumors with a specific hypermethylation pathway, known as CpG island methylator phenotype (CIMP)187. Patients with MSI-H cancers, regardless of their germline or sporadic origins, have distinct pathological and clinical features, including increased numbers of tumor-infiltrating lymphocytes (TILs), frequent poor differentiation and mucinous histology, and proximal colon predominance188. Furthermore, patients with sporadic MSI-H tumors are of particular interest in epidemiological studies, especially with regard to the potential causative factors of cigarette smoking, female gender, and advanced age at diagnosis186. Thus, the defects of MMR gene in CRC could be synthetically lethal with the inhibition of genes controlling the response to oxidative DNA damage, such as DNA polymerase and PTEN-induced putative kinase 1 (PINK1)189. The synthetical lethality of MSH2 or MLH1 and DNA polymerase (POLB or POLG) causes an accumulation of 8-oxoG oxidative DNA lesions that kills cancer cells190. Likewise, inhibition of PINK1 in MMR-deficient colon cancer cell results in an elevation of ROS and the accumulation of both nuclear and mitochondrial oxidative DNA damage that limits cell proliferation191. These findings highlight targeted, mechanism-based therapeutic approaches against CRC.
There is growing evidence linking the gut microbiota, oxidative stress, and inflammation to the genetic and epigenetic alterations observed in CRC184. Advances in high-resolution next-generation sequencing (NGS) technologies have begun to facilitate investigation of the complex etiologic relationship between the microbiome and CRC. NGS analyses on human CRC tissues have enabled identification of microbial compositions related to subtypes of CRC. It is currently a consensus that the microbiota maintains a symbiotic relationship with its host and that it has various functions in both metabolism and immunity192,193. The gut microbiota and the byproducts or metabolites of the microbes therein can trigger inflammation in the colon, which accelerate colon tumorigenesis194. The microbes or products are recognized and bound by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), on CECs and immune cells195. The binding of ligands to TLRs initiates downstream signaling cascades, leading to the activation of NF-κB. Active NF-κB regulates cell survival, proliferation, and inflammatory processes196. Cytokines produced by immune cells might also affect colon tumorigenesis through oncogene activation or gene silencing. For instance, TILs produce large amounts of IL-6, IL-17A, IL-17F, IL-21, IL-22, TNF-a, and other cytokines. These cytokines boost the proliferation of the HIT-29 and DLD-1 CRC cell lines by activating NF-κB and STAT3197. Clinically, MMR-deficient individuals have raised levels of peritumoral lymphocytes. Similar to the case for TILs, raised peritumoral lymphocyte levels are related to a higher risk of developing CRC in MMR-deficient patients198,199. Elevated levels of ROS produced by CECs and immune cells in response to TLR activation can also cause DNA damage and excessive proliferation of CECs200,201. Failure to repair ROS-mediated DNA damage could be an additional contributing mechanism to CRC progression.
Breast and ovary
Breasts and ovaries are crucial female organs closely related to reproduction and sexual characteristics. Each breast is composed of 1525 secretory lobes embedded in subcutaneous adipose tissue. Each lobe is a compound tubular acinar gland. The acini and ducts are lined by an inner layer of cuboidal or columnar epithelial cells and an outer layer of myoepithelial cells. In females, the breasts contain mammary glands that produce and secrete milk to feed infants, while the ovaries are the female gonads. The ovary is composed of an outer cortex and an inner medulla, surrounded by a capsule. Ovaries play important roles in estrogen release, germ cell production, and hormone responses.
According to the Union for International Cancer Control World Cancer Congress in Paris in November 2016, breast cancer is the most commonly diagnosed cancer and the leading cause of cancer mortality among women worldwide, while ovarian cancer accounts for approximately 4% of global cancer cases and deaths among women183,202. Notably, functional mutations in BRCA1 confer lifetime risks of up to 90% for developing breast cancer and 40%50% for ovarian cancer203-205. As a core DNA repair factor, BRCA1 plays crucial roles in DDR processes including cell cycle checkpoint activation and HR repair of DSBs97,206. However, it is unclear why BRCA1 defects tend to induce breast and ovarian cancers rather than other types of cancers. Knowledge of the underlying features of the breast and ovary would serve as a hotspot for understanding tumorigenesis in these two tissues.
One of the most common metabolic materials in the breasts and ovaries is estrogen. Estrogen is produced and secreted by the ovaries, while the breasts are main target tissues207,208. As the major sex hormone of women, estrogen is essential for developing, regulating and maintaining the female reproductive system and secondary sex characteristics. In the human body, estrogen could be normally catabolized by catechol-o-methyltransferase (COMT) for maintaining normal cycling of the hormone209-212. However, when estrogen is present in excess, it can be oxidized into quinone radicals that induce oxidative damage in genomic DNA213-218, and repair of such lesions may require a BRCA1-dependent pathway219. An epidemiological study has also revealed that excessive exposure to estrogen is associated with a higher risk of breast cancer220. Thus, oxidative metabolites of estrogen may be the potential drivers of BRCA1-related breast cancer211 (Figure 2). Other mutations in the DNA repair genes ATM, CHEK2, PALB2, BARD1, BRIP1, RAD50, RAD51C, RAD51D, NBN, FANCM, and MRE11 may also increase the risk of breast and ovarian cancers96,221.

Estrogen oxidation-induced DNA damage and breast cancer. Estrogens (E1: estrone; E2: estradiol) are metabolized through two main pathways, forming 4- and 2-hydroxylated estrogens respectively, known as catechol estrogens (CEs). With further oxidation, CEs are converted to semiquinones (CE-SQs) and then to CE-quinones (CE-Qs), including E-3,4-Q and E-2,3-Q. CE-Q is a type of dangerous ROS, which attacks genomic DNA. This reaction results in the formation of depurinating adducts including 4-OHE-1-N3Ade, 4-OHE-1-N7Gua, and 2-OHE-6-N3Ade, leaving breaks in DNA. If lacking BRCA1, cells cannot repair the DNA damage. Accumulative DNA lesions lead to genomic instability and further breast cancer.
In parallel, estrogen acts on its two receptors, estrogen receptor a (ERa) and estrogen receptor ݠ(ERݩ. The binding of estrogen to its receptors regulates the expression of many genes in a well-organized way222. In particular, it activates the transcription of estrogen-responsive genes, such as genes of growth factors and cell cycle regulating cyclins, and proto-oncogenes, including c-MYC, c-FOS, and HER2/Neu223. The activated genes serve to stimulate cell proliferation and suppress apoptosis224. Estrogen also binds to membrane-bound G-protein-coupled estrogen receptor (GPER, GPR30) to rapidly exert nongenomic effects by activating second messenger systems225. When dysregulated, hyperactive estrogen signaling induces extortionate proliferation, giving rise to DNA damage accumulation and ultimately leading to malignant transformation17. Of note, estrogen signaling has been shown to interact with DDR and DNA repair pathways by regulating key effectors in DDR, including DNA-PKcs, ATM, ataxia telangiectasia and Rad3-related (ATR), BRCA1, BRCA2, and p53, and effector kinases in DNA repair mechanisms226. For instance, ERa may bind DNA-PKcs, stabilizing them and activating DDR through the NHEJ pathway227. ERa downregulates ATM by inhibiting ATM kinase expression via activation of microRNA-18a and microRNA-106a228. Moreover, the ATR protein is negatively regulated by ERa. ERa suppresses ATR activation, ATR-Chk1 signaling at the G2/M checkpoint, and the interaction between DNA Topo II binding protein 1 (TopBP1) and ATR at DNA damage sites. Thus, ER signaling affects not only estrogen-related carcinogenesis but also the processing of other genotoxic insults in estrogen-responsive tissues226.
In the past, DDR defects have been exploited to develop many commonly used chemo- and radio-therapies for breast and ovarian cancer. For example, platinum salts arouse DNA ICLs, the lesions recognized by the DDR and repaired by a combination of HR and NER. These agents can be effective in patients with ovarian carcinomas that commonly harbor defects in HR, especially high-grade serous ovarian cancer204. Today, an alternative approach to traditional therapy is to directly target specific components in DDR pathways through pharmacologic design. Recent years, PARP inhibitors are emerging as a promising new class of chemotherapeutic agents particularly effective against tumors bearing defective BRCA1 and BRCA2229,230. Although nonuniform explanations exist for anticancer mechanism of PARP inhibitors, all the studies highlight the importance of processes of DDR in tissues231-233. The generation of PARP inhibitors can be seen as a milestone of cancer treatment, which not only considerably improves the prognosis of breast and ovarian cancer, but also provides a novel idea for mechanism exploration and target therapy.
Hematopoietic system
The hematopoietic system has been well studied in terms of its development. All blood cell lineages derive from a limited number of hematopoietic stem cells (HSCs) through an actively amplifying progenitor compartment234. As it includes a pool of rapidly proliferating and regenerating cells, the hematopoietic system is also one of the most sensitive and vulnerable systems towards endogenous and exogenous genotoxic insults. HSCs originate from the aorta-gonad-mesonephros (AGM) region, travel to certain anatomic sites, including the placenta, spleen, liver, and lymph nodes of the developing fetus, and ultimately localize to the bone marrow cavity during late embryogenesis. After birth, fetal HSCs gradually develop into quiescent adult HSCs and remain in bone marrow throughout their lifetime.
It has been indicated that HSCs and other tissue-specific stem cells should have cellular and genomic protective mechanisms to maintain their functional potential throughout their lifespans. Indeed, increasing evidence suggests that HSCs are armed with multiple protective properties responsible for preserving their activity. For instance, the expression of telomerase and the dormant nature of HSCs help to prevent the uncapping of telomeres or the introduction of aberrations during replication235-237. ROS in HSCs are maintained at relatively low levels because of the low metabolic activity in these cells238. It is noteworthy that ATM-deficient HSCs contain increased level of ROS and display an overall function decline over time, resulting in eventual hematopoietic failure239. On clinic, isocitrate dehydrogenase-1 gene (IDH1) deficiency is a common driver of acute myeloid leukemia (AML). Mechanically, mutant IDH1 downregulates the expression of the DNA damage sensor ATM by altering its histone methylation. Decreased ATM results in impaired DNA repair and increased sensitivity to DNA damage, which reduces HSC self-renewal240.
Notably, the functions of prosurvival genes in Bcl-2 family and of p53-mediated DDR processes have been emphasized in HSCs for IR injury repair241,242. In quiescent HSCs, heightened expression of prosurvival genes, such as Bcl2, Bcl-Xl, Mcl1, and A1, inhibits cell death prompted by p53. In these cases, the proapoptotic genes Bax, Noxa, and Puma permit p21 to participate in a transient growth-arrest response for DNA repair243. Different from quiescent HSCs that employ the error-prone NHEJ pathway upon DNA damage, proliferating HSCs use the high-fidelity HR pathway to repair DSBs243. A study involving short-term culture of isolated quiescent HSCs revealed that γ-H2AX-marked DSBs are markedly reduced after HSCs enter the cell cycle, indicating that proliferating HSCs repair DSBs better than quiescent HSCs244. Also, this may be the reason why patients who undergo chemotherapy or radiotherapy tend to develop leukemias and lymphomas because the use of error-prone DNA repair in quiescent HSCs would potentially cause a large number of DNA lesions. The major outcome of the DDR in fetal HSCs is an increased level of apoptosis and cell elimination promoted by apoptosis-stimulating of p53 protein 1 (ASPP1). In contrast, adult HSCs demonstrate a very different response to IR, with DNA repair and overt cell survival being the principal outcomes243. This difference in response might be attributable to the different roles of HSCs during these stages. During embryogenesis, the vital goal of hematopoiesis is to amplify the stem cells and protect their genomic integrity to establish an HSC pool. This ensures blood homeostasis for the lifetime of the organism. In adults, however, the major function of the HSC compartment is to preserve blood homeostasis and quick response to hematopoietic needs associated with conditions such as infection and blood loss245. Moreover, HSCs are more resistant to IR than their downstream progeny, which prevents the exhaustion of the HSC pool246.
Due to its vigorous, sensitive, and amorphous properties, the hematopoietic system generally responses well to DDR inhibition treatments. Still, because of the significant diversity lying in blood cells of different stages and different types of hematopoietic cancers, DDR inhibitors vary in clinical indications. In a recent study, scientists find that a novel ATR kinase inhibitor AZD6738 leads to an accumulation of unrepaired DNA damage in p53- or ATM-defective chronic lymphocytic leukemia (CLL) cells. The synthetic lethality is achieved by mitotic catastrophe caused by defective cell cycle checkpoints, resulting in a selective cytotoxicity to both p53- and ATM-defective CLL cells247. Another interesting observation comes from AML, myelodysplastic syndrome, juvenile/chronic myelomonocytic leukemia, and T-cell acute lymphoblastic leukemia harboring KRAS mutations. The oncogenic KRAS causes a shift in DSB repair from the canonical-NHEJ pathway to a preferred use of the more error-prone alternative-NHEJ (alt-NHEJ) pathway248. Thus, targeting the components of the alt-NHEJ pathway could sensitize KRAS-mutated leukemic cells to standard chemotherapeutics and represents a promising approach for inducing synthetic lethality in cancer cells.
Conclusions
DNA damage and repair are inevitable and indispensable cellular processes that serve prominently to determine the fates of cells, organs, systems, and even the whole body. When DNA damage occurs, cells respond correspondingly. Though general mechanisms and DDR pathways are shared, the fates of cells greatly rely on tissue-specific processes. Aside from the tissue-specific expression of DDR conserved genes, epigenetic modulation, cytokines, hormones and cellular niches also precisely regulate cell fate in a tissue-specific manner. The combination of intrinsic properties and regulatory factors accounts for the specificity and precision of DDR.
DDR serves as not only a guardian of genomic integrity, but also an oncogene-inducible physiological barrier against tumorigenesis and early tumor progression. Mutations in different DDR pathways tend to arouse very different clinical events including tissue-specific tumorigenesis described above (Table 1). In the past decade, plenty of promising DDR inhibitors have been developed on clinic. The inhibition of DDR in a specific tissue or microenvironment preferentially kills cancer cells while spares normal cells, thus providing significant patient benefits over conventional chemotherapies249. Advances in NGS technologies would also facilitate the identification of patient subgroups or particular cancer types with DDR defects, and better instruct the personalized therapy250.
Tissue specific DDR and tumorigenesis
Still, the relationship between reported mutations and dysfunctions of the DDR proteins have yet to be defined. Comprehensive knowledge regarding the tissue-specific DNA damage and repair processes in different species, developmental stages, and cell cycle phases remains much limited. Prompt repair and cellular survival may not be the ultimate goal for each cell. Instead, tissues with distinct features and functions may prioritize different processes to maximize the overall wholesome state of the body. For instance, altruistic suicide of intestinal stem cells in response to DNA damage could prevent the transmission of precancerous mutations, which potentially explains the rarity of intestinal cancers compared to colonic neoplasms245. Why are certain pathways more sensitive and responsive towards DNA damage in some tissues than in others? What are the decisive elements among tissues? How do certain mutations or functional defects in DDR gradually develop into adverse consequences? The answers to these questions will provide profound insights into the mechanisms underlying the progression of tumorigenesis and novel clues for the development of effective anticancer treatments.
Acknowledgements
We are grateful to Dr. Ning Zhang for the insightful comments and suggestions on the manuscript. This work was supported by the National Natural Science Foundation of China (Grant No. 81622035, 81672610, and 81521002).
Footnotes
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received March 5, 2019.
- Accepted April 30, 2019.
- Copyright: © 2019, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.
- 41.
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.↵
- 69.↵
- 70.
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.
- 80.
- 81.
- 82.
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.
- 126.↵
- 127.↵
- 128.↵
- 129.
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.
- 136.
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.
- 168.↵
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.↵
- 174.↵
- 175.↵
- 176.↵
- 177.↵
- 178.↵
- 179.↵
- 180.↵
- 181.
- 182.↵
- 183.↵
- 184.↵
- 185.↵
- 186.↵
- 187.↵
- 188.↵
- 189.↵
- 190.↵
- 191.↵
- 192.↵
- 193.↵
- 194.↵
- 195.↵
- 196.↵
- 197.↵
- 198.↵
- 199.↵
- 200.↵
- 201.↵
- 202.↵
- 203.↵
- 204.↵
- 205.↵
- 206.↵
- 207.↵
- 208.↵
- 209.↵
- 210.
- 211.↵
- 212.↵
- 213.↵
- 214.
- 215.
- 216.
- 217.
- 218.↵
- 219.↵
- 220.↵
- 221.↵
- 222.↵
- 223.↵
- 224.↵
- 225.↵
- 226.↵
- 227.↵
- 228.↵
- 229.↵
- 230.↵
- 231.↵
- 232.
- 233.↵
- 234.↵
- 235.↵
- 236.
- 237.↵
- 238.↵
- 239.↵
- 240.↵
- 241.↵
- 242.↵
- 243.↵
- 244.↵
- 245.↵
- 246.↵
- 247.↵
- 248.↵
- 249.↵
- 250.↵
In this issue






{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Detecting haplotype-specific transcript variation in long reads with FLAIR2
- Extended family with an inherited pathogenic variant in polymerase delta provides strong evidence for recessive effect of proofreading deficiency in human cells
- Tissue- and sex-specific DNA damage tracks aging in rodents and humans
- Anti-tumor pharmacology of natural products targeting mitosis
- Harnessing DSB repair to promote efficient homology-dependent and -independent prime editing