

Abstract
Objective Heat shock factor 1 (HSF1), a transcriptional regulator of heat shock proteins (HSPs), is an attractive therapeutic target for cancer. However, only a few HSF1 inhibitors have been identified so far.
Methods The mRNA and protein levels of HSF1, HSPs, cleaved PARP, and phosphorylated HSF1 were examined by real-time PCR and Western blot. Forced expression, RNA interference, and immunofluorescence assay were used for mechanistic studies. Cell viability and apoptosis were measured by WST-8 assay and flow cytometry, respectively. Xenograft studies were performed in nude mice to evaluate the effect of dorsomorphin and an HSP90 inhibitor on tumor growth.
Results Dorsomorphin suppressed multiple stimuli-induced and constitutive HSPs expression in cancer cells. Mechanistic studies revealed that dorsomorphin reduced heat-induced HSP expression independent of adenosine monophosphate activated protein kinase. Dorsomorphin reduced heat-stimulated HSF1 Ser320 phosphorylation and nuclear translocation, as well as resting nuclear HSF1 levels in cancer cells. Dorsomorphin induced cancer cell apoptosis by inhibiting HSF1 expression. A structure-activity study revealed that the 4-pyridyl at the 3-site of the pyrazolo [1, 5-a]pyrimidine ring is critical for the anti-HSF1 activities of dorsomorphin. Dorsomorphin sensitized cancer cells to HSP90 and proteasome inhibitors and inhibited HSP70 expression induced by these inhibitors in vitro. In tumor-bearing nude mice, dorsomorphin enhanced HSP90 inhibitor-induced cancer cell apoptosis, tumor growth inhibition, and HSP70 expression.
Conclusions Dorsomorphin is an HSF1 inhibitor. It induces cancer cell apoptosis, sensitizes cancer cells to both HSP90 and proteasome inhibitors, and suppresses HSP upregulation by these drugs, which may prevent the development of drug resistance. Hence, dorsomorphin and its derivates may serve as potential precursors for developing drugs against cancer.
keywords
Introduction
Heat shock factor 1 (HSF1) is a master transcriptional regulator of heat shock response, which protects organisms against various environmental and pathological stimuli. Under resting conditions, inactive HSF1 monomers are mainly located in the cytoplasm in a complex with inhibitory proteins, such as heat shock proteins (HSPs; HSP40, 70, and 90). Under stress conditions, HSF1 dissociates from inhibitory proteins, trimerizes, phosphorylates, and translocates to the nucleus, where it binds to heat shock elements in the promoter regions of HSP genes and initiates HSP mRNA expression. HSPs are important chaperone proteins that are essential for cell survival under conditions of cell stress. HSF1 has been reported to be overexpressed or activated in various types of human cancers1-3. The elevation of HSF1 expression in cancer tissues is correlated with increased metastatic potential, resistance to therapy, and poor prognosis1,4. Increasing evidence has demonstrated that HSF1 is critically involved in tumorigenesis5-7, angiogenesis8, and metastasis9,10. HSF1 promotes cancer development and progress through HSP-dependent and -independent mechanisms. HSF1 promotes tumor progression by upregulating HSP expression. Numerous studies have demonstrated that HSP90, 70, 40, and 27 play critical roles in tumor growth and metastasis11-15, and many HSP90 inhibitors are being tested in preclinical and clinical trials. In addition to HSPs, HSF1 upregulates the expression of various proteins that are involved in protein translation, cell cycle regulation, signal transduction, metabolism, adhesion, and cell survival and contributes to cancer cell proliferation, angiogenesis, migration, and invasion1,2,8,16. In addition to contributing to tumorigenesis, HSF1 has been reported to contribute to cancer drug resistance. The proteasome inhibitors and HSP90 inhibitors enhance proteotoxic stress in cancer cells, causing cell cycle arrest and apoptosis. These compounds are presently being tested in clinical trials. However, they induce drug resistance via the activation of HSF1 and the induction of numerous cytoprotective proteins, including HSP70 and HSP90 family members that ameliorate proteostatic damage17-21. In light of its multifaceted roles in oncogenesis and drug resistance, HSF1 is being considered as an attractive therapeutic target for cancer.
It has been reported that HSF1 knockout suppressed chemically induced liver carcinoma and skin cancer6,22. HSF1 knockdown has a minimal effect on normal cell viability but significantly impairs the proliferation of cancer cells22. Several HSF1 inhibitors have been reported to induce cancer cell apoptosis, inhibit tumor growth, and enhance the anti-cancer activity of HSP90 inhibitors2,4. Therefore, identification of new HSF1 inhibitors may not only provide therapeutic drugs against cancer but also prevent drug resistance induced by other anti-cancer drugs.
Dorsomorphin {6-[4-(2-piperidin-1-ylethoxy)phenyl]-3-pyridin-4-ylpyrazolo (1, 5-a)pyrimidine}, also termed compound C, is widely used as an inhibitor of adenosine monophosphate activated protein kinase (AMPK) in metabolic research23. Dorsomorphin has been reported to induce cancer cell apoptosis24-27. Several studies have reported that the proapoptotic effect of dorsomorphin is independent of its inhibition of AMPK25,26; however, the underlying mechanisms are not completely understood. Our previous study found that during heat shock response, HSP70 expression was upregulated by phosphatase 2A (PP2A)-mediated AMPK inhibition28. However, treatment with the AMPK inhibitor dorsomorphin inhibited heat stress-induced HSP70 expression. In this study, we identified dorsomorphin as an HSF1 inhibitor that reduced stress-induced and constitutive HSP expression by inhibiting HSF1 nuclear translocation and reduced nuclear HSF1 levels. We also found that dorsomorphin induced cancer cell apoptosis through inhibiting HSF1. We then studied the structure-activity of dorsomorphin in inhibiting heat-stimulated HSP70 expression and the effect of dorsomorphin derivatives on heat shock response and cancer cell viability. We further studied the effect of dorsomorphin in combination with HSP90 or proteasome inhibitor on cancer cell viability, tumor growth, and inhibitor-induced HSP upregulation. Our findings suggest that dorsomorphin and its derivatives may serve as potential therapeutic agents against cancer through targeting HSF1.
Materials and methods
Cell culture and chemicals
HCT116, HeLa, and PC-3 cells were obtained from American Type Culture Collection (Rockville, MD, USA). Huh7 cells were obtained from Cell Bank of Chinese Academy of Sciences (Shanghai, China). HCT116 cells were cultured in McCoy’s 5A medium. HeLa, PC-3, and Huh7 cells were cultured in Dulbecco’s modified Eagle’s medium. All media contained 10% fetal bovine serum and 10 units/mL penicillin, as well as 10 units/mL streptomycin. For heat stress, the cells were exposed to 42°C in a humidified atmosphere with 5% CO2. Dorsomorphin, Dorsomorphin·2HCl, MG132, and 17-AAG were obtained from Selleck Chemicals (Houston, TX, USA). Dorsomorphin derivatives were purchased from ChemPartner (Shanghai, China).
Cell transfection and RNA interference
Huh7 cells were split and cultured for 24 h, and then were transfected with pcDNA3.1 or pcDNA3.1-HSF1 using Lipofectamine 3000 (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. After 24 h, the cells were examined for HSF1 expression by Western blot or treated with dorsomorphin for another 12 h and examined for HSP70 and cleaved PARP expression by Western blot.
AMPK expression in HeLa cells was inhibited by RNA interference. Briefly, HeLa cells were transfected with 30 nM siRNA against AMPKα (siAMPKα) or scrambled siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. AMPKα expression was examined by Western blot after 48 h.
RNA isolation and quantitative real-time PCR (qPCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Waltham, MA, USA) and treated with RNase-free DNase to degrade contaminating genomic DNA according to the manufacturer’s instructions. cDNA was synthesized from 2 μg RNA with M-MLV reverse transcriptase and oligo (dT) primers. qPCR was performed by using an ABI Prism 7,900 sequence detector system (Applied Biosystems, Waltham, MA, USA). The assays were initiated for 5 min at 95°C, 40 cycles of 15 sec at 94°C and 1 min at 60°C. Amplification of the target cDNA was normalized to β-actin expression. The relative expression of the target mRNA was calculated using the 2–ΔΔCT method. The primer sequences used for qRT-PCR are listed in Supplementary Table S1.
Western blot
The cells were lysed in ice-cold lysis buffer containing protease inhibitors (Calbiochem, Darmstadt, Germany). The supernatant was collected by centrifugation and protein concentration was measured using the BCA kit (Beyotime Biotechnology, Shanghai, China). Western blot was conducted according to standard protocol. The following primary antibodies were used: antibodies against HSP70, HSF1, AMPKα, and cleaved PARP (Cell Signaling Technology, Danvers, MD, USA), and phosphorylated HSF1 (S303+S307, S320) (Abcam, Cambridge, UK). β-actin (Sigma-Aldrich, St. Louis, MO, USA), α-tubulin, and lamin B1 (Proteintech Group Inc., IL, USA) were used as loading controls. Horseradish peroxidase (HRP)-conjugated Affinipure Goat Anti-Mouse IgG (H+L) and HRP-conjugated Affinipure Goat Anti-Rabbit IgG (H+L) were used as secondary antibodies. The target proteins were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA). Representative blots from at least three independent experiments are shown.
To detect HSF1 levels in the cytoplasm and nucleus via Western blot, nuclear and cytoplasmic protein extraction was performed as previously described29 with the modification that solution B was replaced with lysis buffer.
The Western blot bands were quantified with ImageJ software.
Cell viability assay
Cell viability was measured by using Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Inc., Kumamoto, Japan), which detects dehydrogenase activity. Briefly, the cancer cells were seeded into 96-well plates (5,000 cells/well), cultured in an incubator for 24 h, and treated with different concentrations of dorsomorphin or its derivatives for 24 h. CCK-8 solution (10 μL) was then added to each well of the plate, and the cells were incubated at 37°C for 2–3 h in the dark. The absorbance at 450 nm was measured using a microplate reader (PerkinElmer, Boston, MA, USA).
Immunofluorescence analysis
HeLa cells growing on coverslips were treated with dorsomorphin for 30 min followed by exposure to 42°C for 1 h. Cells were then washed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde for 15 min at room temperature. After 3 washes with PBS, the cells were treated with blocking buffer (1% bovine serum albumin/0.3% Triton X-100 in PBS) for 1 h at room temperature, incubated with primary antibody against HSF1 overnight at 4°C, and washed with PBS. The bound primary antibodies were detected by incubating with FITC-tagged secondary antibody for 1 h at room temperature. After washing with PBS, the DNA was stained with DAPI (Keygen Biotech, Nanjing, China) for 5 min. The staining pattern was analyzed using a laser confocal microscope.
Apoptosis assay
Cell death was assessed using the Annexin V-FITC/PI (propidium iodide) Apoptosis Detection Kit (KGA108; Keygen Biotech, Nanjing, China). HeLa cells were treated with control medium, dorsomorphin, 17-AAG, MG132, 17-AAG + dorsomorphin, or MG132 + dorsomorphin for 24 h. After staining with annexin V-FITC/PI, flow cytometric analysis was performed using a flow cytometer (Beckman Coulter Inc., Brea, CA, USA).
Tumor xenograft mouse model and treatment
Five-week-old male athymic BALB/c nude mice were purchased from Shanghai Slac Laboratory Animal Co. (Shanghai, China) and allowed to acclimate for one week in a specific pathogen-free animal facility. HCT116 cells [4 × 106 cells suspended in 0.15 mL mixture containing an equal volume of McCoy's 5A medium and Matrigel (BD Biosciences, New Jersey, NJ, USA)] were injected subcutaneously into the right flanks of mice. The tumor size was measured with a vernier caliper every two days. The tumor volume was calculated by the following formula: tumor volume = 1/2[length (mm) × width (mm)2]. When the tumors grew to an average volume of 250 mm3, the mice were randomly assigned into four groups (n = 6–9 per group) as follows: control, 17-AAG, dorsomorphin and dorsomorphin + 17-AAG. Dorsomorphin·2HCl was dissolved in PBS; 17-AAG was dissolved in 5% DMSO containing corn oil. Dorsomorphin (10 mg/kg body weight) was administered twice per week through intratumoral injection; 17-AAG (50 mg/kg body weight) was intraperitoneally injected thrice per week. The control group was treated with the solvents for dorsomorphin and 17-AAG, the dorsomorphin group was administered dorsomorphin and the solvent for 17-AAG, and the 17-AAG group was administered 17-AAG and the solvent for dorsomorphin. The animal research was reviewed and approved by the Institutional Animal Care and Use Committee of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
Statistical analysis
Data were presented as mean ± standard deviation (SD) or mean ± standard error (SE). Statistical differences between the test and control groups were analyzed by Student’s t-test. A P-value of less than 0.05 was considered statistically significant.
Results
Dorsomorphin inhibits HSP expression under various conditions
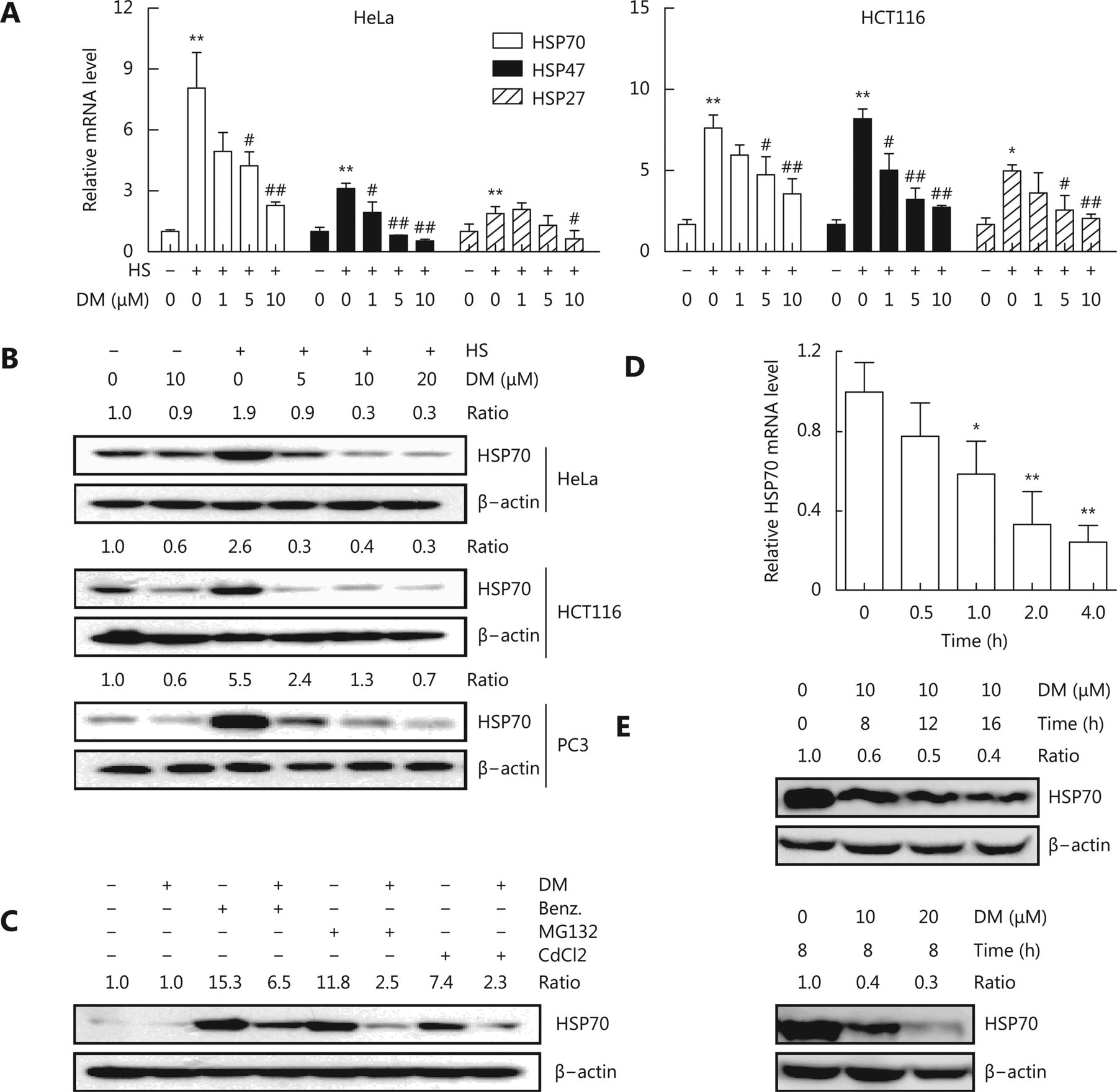
We first examined the effect of dorsomorphin on the expression of HSPs in response to heat stress. Exposure of HeLa and HCT116 cells to 42°C for 1 h significantly induced the mRNA expression of HSPs, including HSP70, HSP47, and HSP27 (Figure 1A). Pretreatment of these cells with dorsomorphin inhibited heat stress-induced HSP expression in a concentration-dependent manner (Figure 1A). Next, we evaluated the effect of dorsomorphin on heat stress-induced protein expression of HSPs, especially that of HSP70, which plays a critical role in cancer development and is a therapeutic target for cancer. We observed that dorsomorphin suppressed heat-induced HSP70 protein expression in different types of cancer cells (Figure 1B). We found that in addition to inhibiting heat stress-induced HSP expression, dorsomorphin inhibited HSP70 expression in response to other HSP inducers, such as the HSP90 inhibitor benzisoxazole, the proteasome inhibitor MG132 and CdCl2 (Figure 1C). We further evaluated the effect of dorsomorphin on HSP70 expression under resting conditions and observed that dorsomorphin downregulated HSP70 expression at both mRNA and protein levels in cancer cells (Figure 1D and 1E). Altogether, we demonstrated that dorsomorphin inhibits HSP expression in multiple cancer cell lines under various conditions.
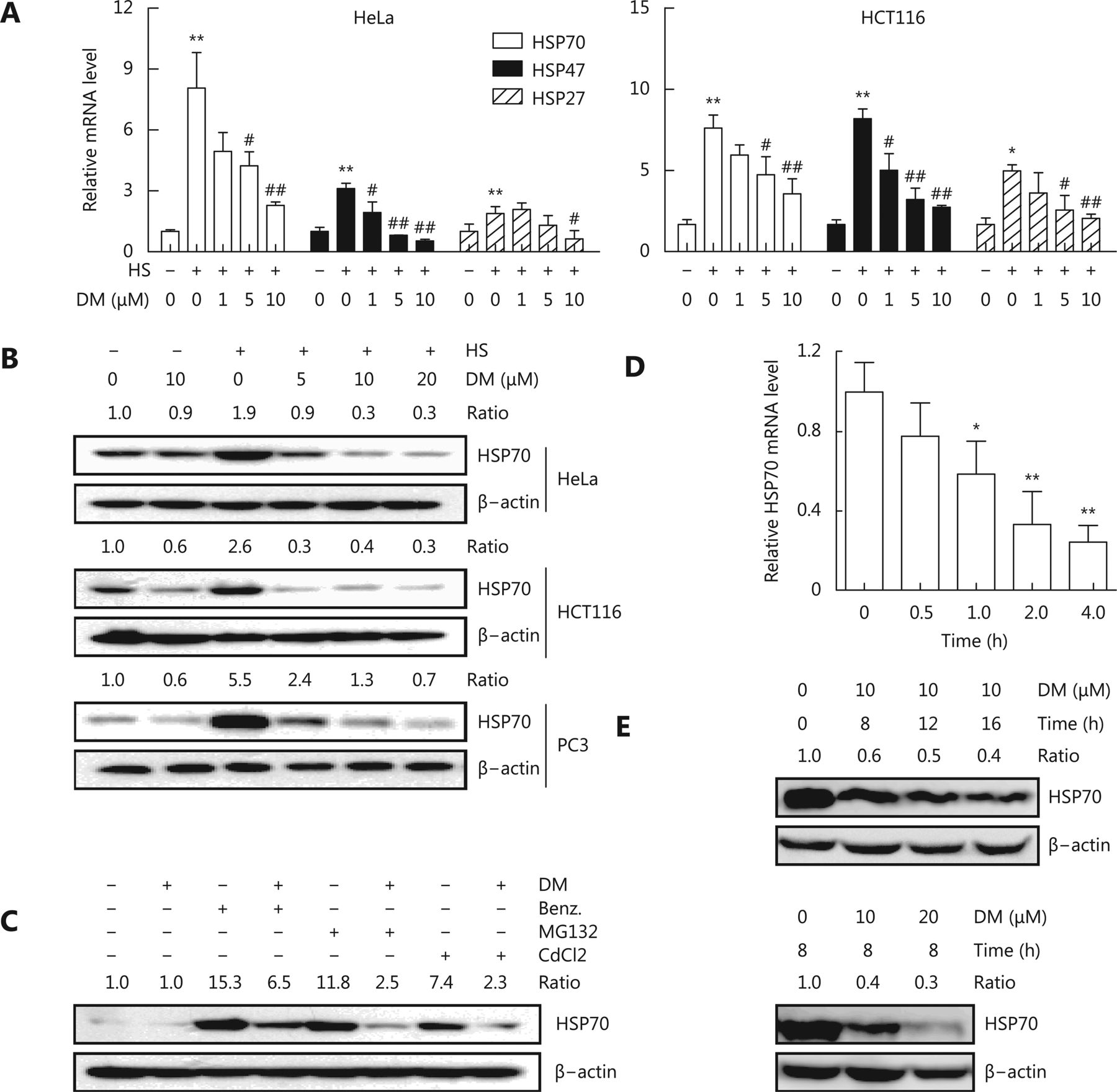
Dorsomorphin (DM) inhibits heat shock protein expression. (A-B) Cancer cells pretreated with or without different concentrations of DM for 30 min were exposed to 42 °C (heat stress, HS) for 1 h. The mRNA levels of heat shock proteins (HSPs) were examined by qRT-PCR (A), and the protein levels of HSP70 were examined by Western blot after recovery at 37 °C for 5 h (B). (C) HeLa cells pretreated with 10 μM DM for 30 min were stimulated with 0.5 μM HSP90 inhibitor benzisoxazole (Benz.), 20 μM MG132, or 50 μM CdCl2 for 8 h and examined for HSP70 expression by Western blot. (D-E) HCT116 cells were incubated with 10 μM DM for different periods of time (D, E upper panels) or with different concentrations of DM for 8 h (E lower panels), and HSP70 expression was examined at mRNA (D) or protein level (E). Data shown represent the mean ± SD, n = 3. *P < 0.05, ** P < 0.01, compared with cells unexposed to heat (A) or DM (D). #P < 0.05, ##P < 0.01 compared with cells exposed to heat alone (A). B, C and E are representative images of three independent experiments with similar results.
Dorsomorphin inhibits HSF1 nuclear accumulation
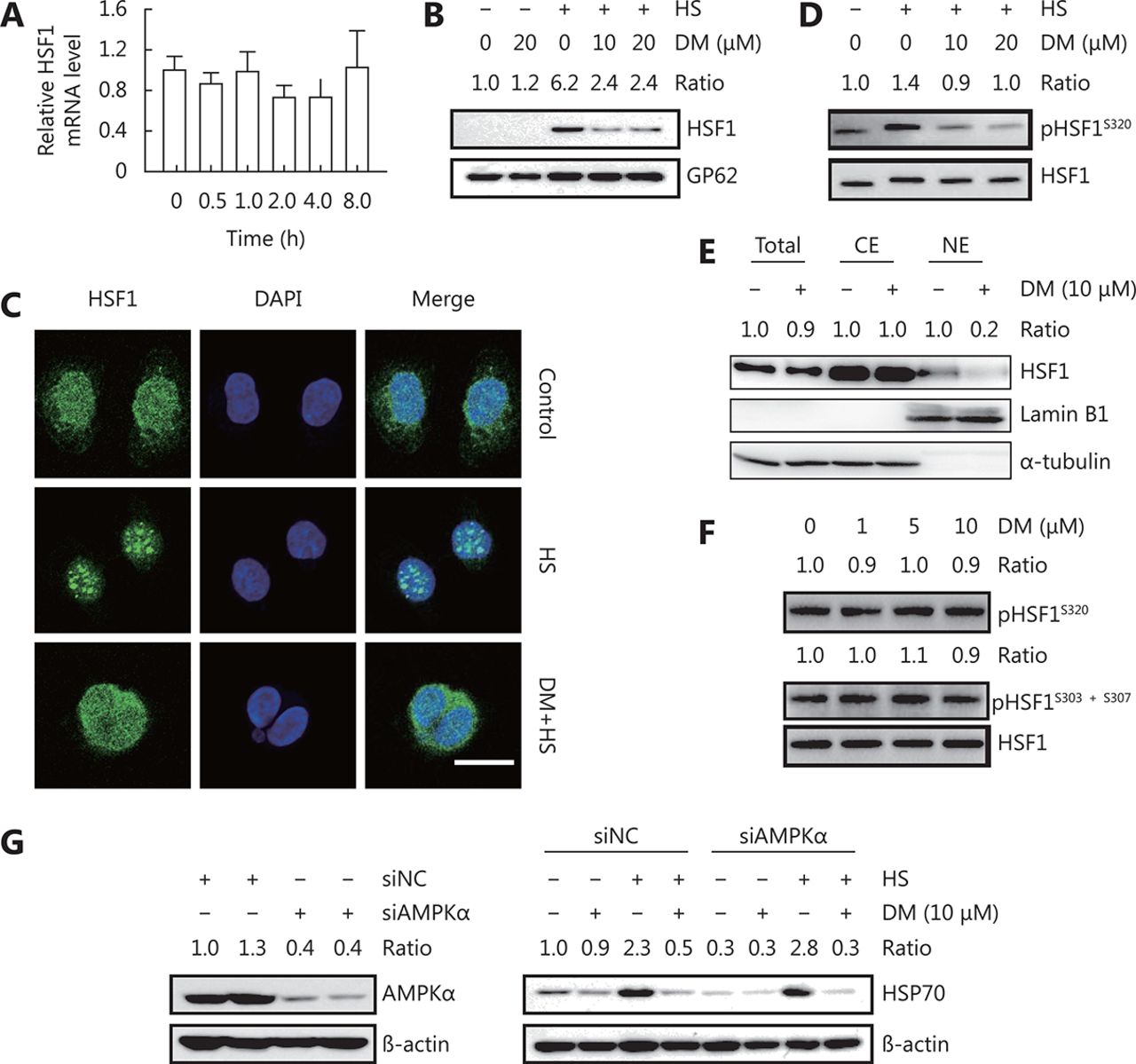
As dorsomorphin could inhibit HSP expression induced by various stimuli, we speculated that it might target common molecule(s) in heat shock response. HSF1 is a major regulator of heat shock response. Upon heat shock or proteotoxic stress, HSF1 dissociates from HSP90, HSP70, and HSP40, trimerizes, phosphorylates, translocates to the nucleus, and binds to the heat shock element at the promoters of its target genes to modulate their transcription. We evaluated the effect of dorsomorphin on HSF1 expression and activation during heat shock response and observed that treatment of cancer cells with 10 μM dorsomorphin had no significant effect on HSF1 expression at both mRNA and protein levels (Figure 2A and 2D). Exposure of cells to 42°C induced HSF1 nuclear accumulation (Figure 2B) and nuclear HSF1 granule formation (Figure 2C). Pretreatment of the cells with dorsomorphin markedly inhibited heat-induced HSF1 nuclear translocation (Figure 2B) and suppressed HSF1 granule formation in the nuclei (Figure 2C). It has been reported that phosphorylation of HSF1 at Ser320 and Ser419 facilitates HSF1 nuclear translocation30,31. We found that pretreatment of HeLa cells with dorsomorphin reduced HSF1 Ser320 phosphorylation induced by heat stress (Figure 2D), indicating that dorsomorphin may inhibit heat-induced HSF1 nuclear translocation through suppressing HSF1 Ser320 phosphorylation. Furthermore, we found that dorsomorphin could reduce nuclear HSF1 levels in HCT116 cells under unstressed conditions (Figure 2E). However, dorsomorphin had no effect on HSF1 phosphorylation at Ser320 (Figure 2F). It has been reported that phosphorylation of HSF1 at Ser303 and Ser307 promotes nuclear HSF1 export3,32. We investigated whether dorsomorphin could modulate the phosphorylation of these amino acid residues in HSF1 and obtained negative results (Figure 2F). These results indicate that dorsomorphin inhibits HSP expression by blocking HSF1 nuclear translocation.
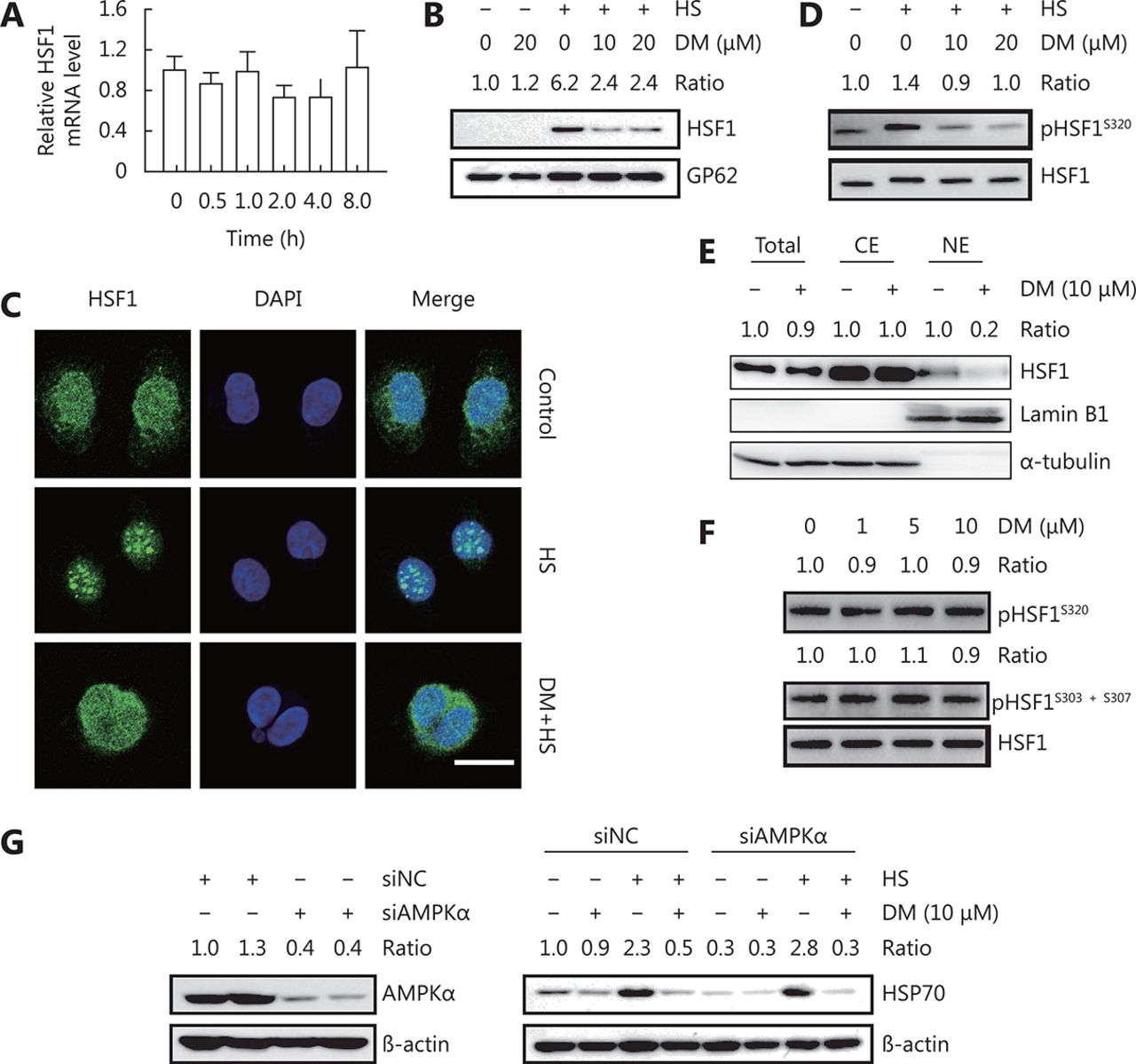
DM inhibits HSF1 nuclear translocation. (A) HCT116 cells were incubated with 10 μM DM for different periods of time, and HSF1 mRNA levels were examined by qRT-PCR. (B-D) HeLa cells pretreated with DM for 30 min were exposed to 42°C (heat stress, HS) for 1 h. Nuclear and total HSF1 was examined by Western blot (B) and immunofluorescence staining, scale bar = 20 μM (C), pHSF1S320 was examined by Western blot (D). (E) HCT116 cells were treated with DM for 16 h. HSF1 levels in total cell lysate, cytoplasmic extract (CE), or nuclear extract (NE) were examined by Western blot analysis, (F) pHSF1S320 and pHSF1S303+S307 were detected by Western blot analysis (G) HeLa cells transfected with AMPKα siRNA or control siRNA for 48 h were examined for AMPKα expression by Western blot (left panels) or treated with 10 μM DM for 30 min followed by exposure to 42°C for 1 h, recovery at 37°C for another 5 h and examined for HSP70 expression by Western blot (right panels). The images in B-G are representative results of three independent experiments with similar results.
Because dorsomorphin has been widely used as an inhibitor of AMPK, we evaluated if AMPK is involved in the inhibitory effect of dorsomorphin on heat shock response. We found that knockdown of AMPKα had no effect on heat stress-induced HSP70 expression or dorsomorphin-induced inhibition of HSP70 expression (Figure 2G), indicating that AMPK may not be involved in the inhibition of heat shock response by dorsomorphin.
Collectively, our studies demonstrate that dorsomorphin inhibits HSP expression through inhibiting HSF1 nuclear translocation and reducing nuclear HSF1 levels.
Overexpression of HSF1 reversed dorsomorphin-induced apoptosis
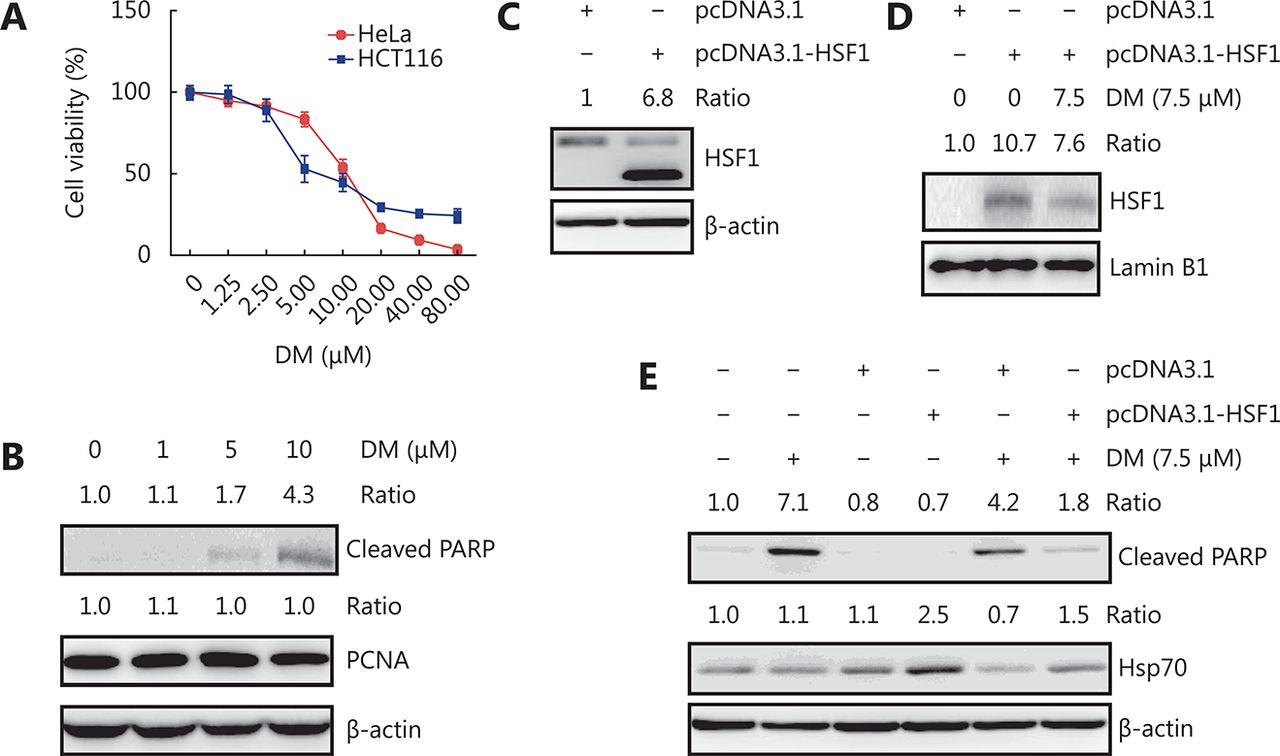
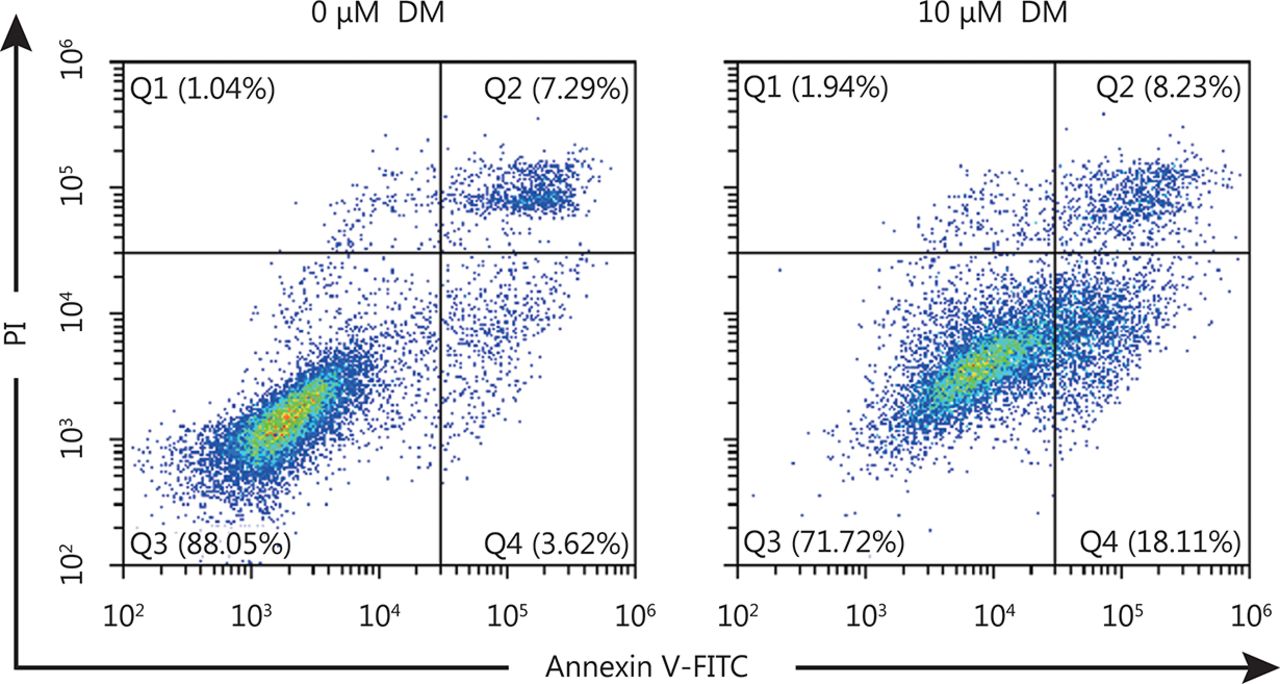
Dorsomorphin has been reported to induce cancer cell apoptosis. We observed that dorsomorphin suppressed the viability of HeLa and HCT116 cells with IC50 values of 10.71 and 11.34 μM, respectively (Figure 3A). Treatment of HeLa cells with dorsomorphin increased the cleaved PARP levels and the percentages of annexin V+ cells, but had no effect on proliferating cell nuclear antigen (PCNA) expression, indicating that dorsomorphin induces apoptotic cell death but has no effect on cell proliferation (Figure 3B and Supplementary Figure S1). To determine whether HSF1 is involved in dorsomorphin-induced cancer cell apoptosis, we transfected Huh7 cells with constitutively active HSF1-expressing plasmids. We observed that overexpression of HSF1 in cancer cells increased nuclear HSF1 levels , which was reduced by dorsomorphin (Figure 3C and 3D), HSF1 overexpression reversed dorsomorphin-induced apoptosis and inhibition of HSP70 expression (Figure 3E). These results indicate that dorsomorphin induces cancer cell apoptosis through inhibiting HSF1-mediated HSP70 expression.
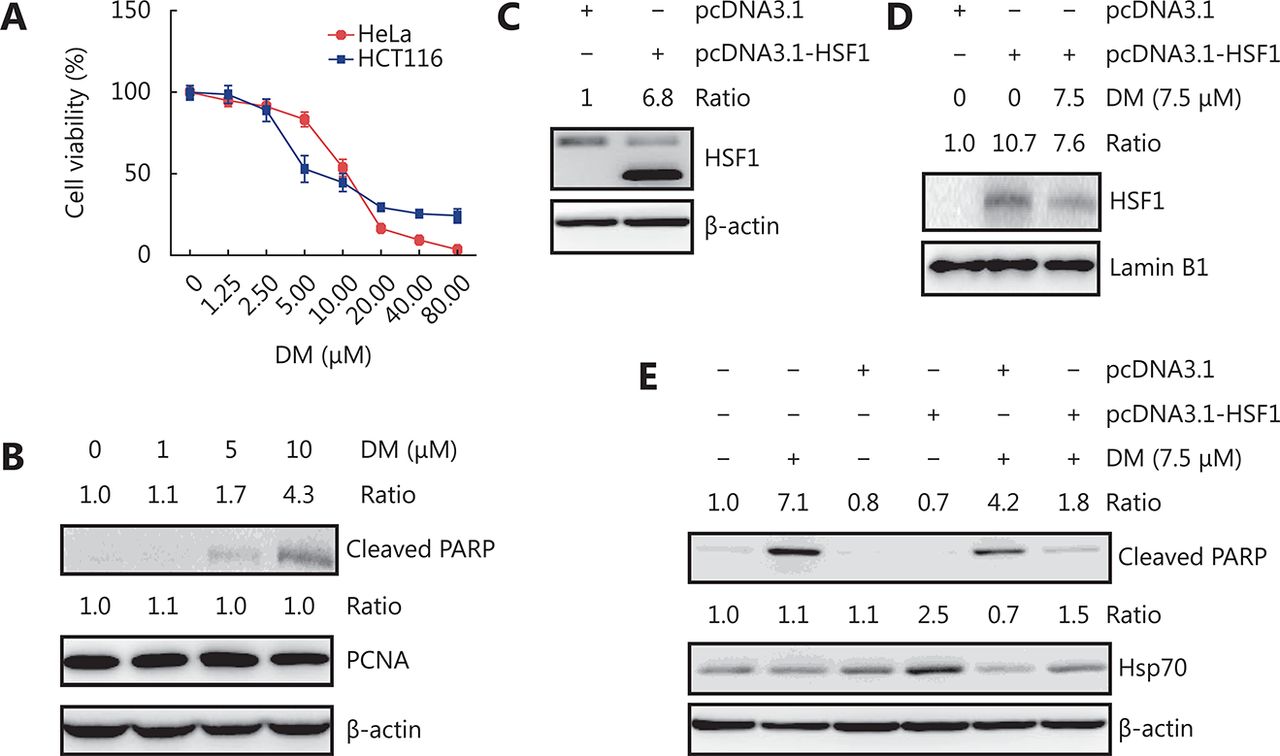
DM-induced apoptosis is reversed by HSF1 overexpression. (A-B) Cancer cells were treated with different concentrations of DM for 24 h, cell viability was examined with CCK-8 (A), cleaved PARP and PCNA were examined by Western blot in HeLa cells (B). Huh7 cells transfected with constitutively active HSF1-expressing plasmid pcDNA3.1-HSF1 or control vector pcDNA3.1 for 48 h were examined for HSF1 expression by Western blot (C) or treated with 7.5 μM DM for 12 h, followed by detection of nuclear HSF1 (D) or HSP70 and cleaved PARP in total cell lysate by Western blot (E). Images in B-E are representative results of three independent experiments.
Dorsomorphin derivatives inhibit heat shock response and induce cancer cell apoptosis
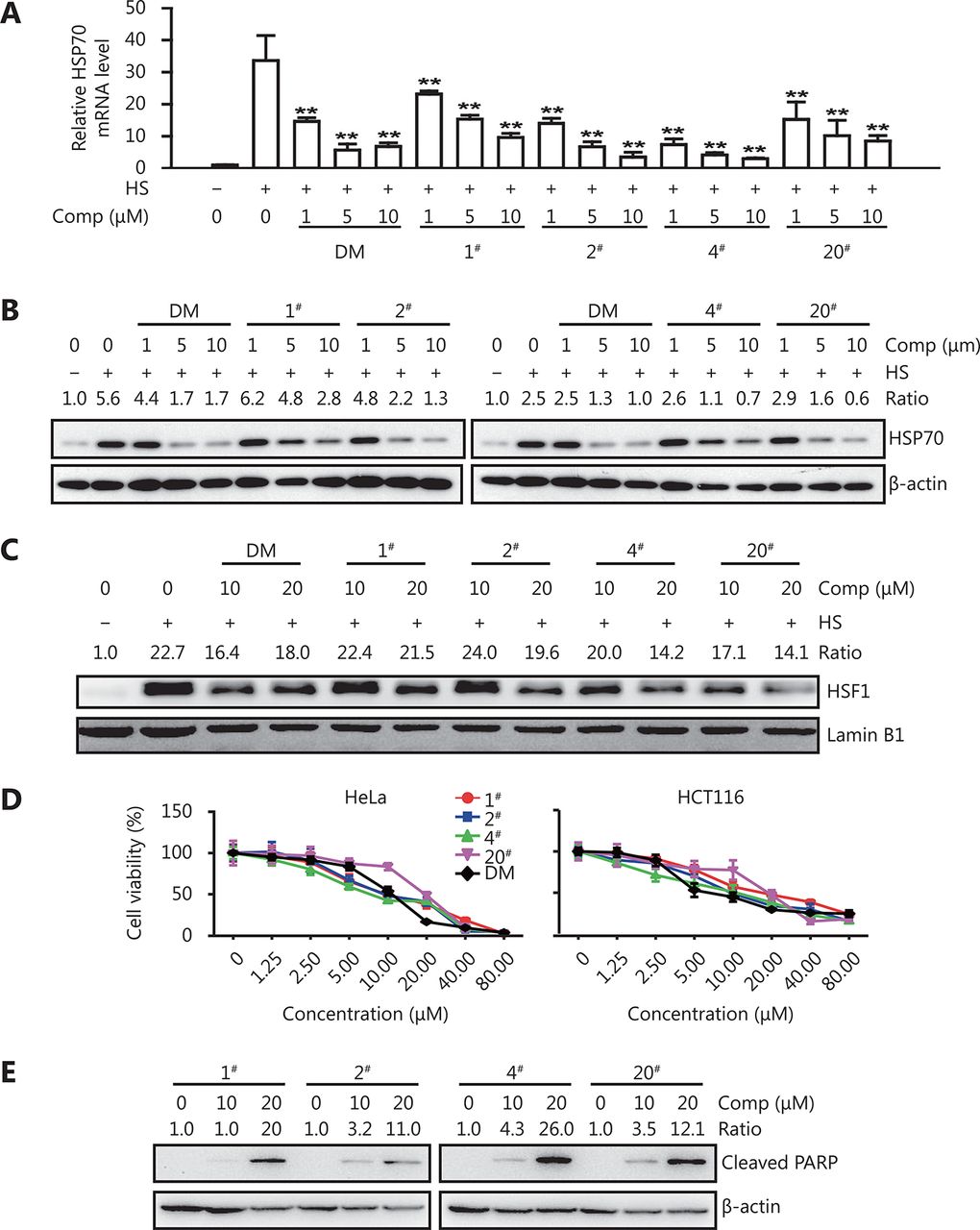
To study the structure-activity relationship of dorsomorphin to inhibit heat shock response and induce cancer cell apoptosis, we synthesized 20 dorsomorphin derivatives by modifying the R1 and R2 moieties, as shown in Table 1. The 4-pyridyl (R1) at the 3-site of the pyrazolo [1, 5-a] pyrimidine scaffold of dorsomorphin was substituted with 3-pyridyl, 5-(2-methoxy) pyridyl, or 4-isoquinolyl. The piperidyl (R2) in the [4-(2-piperidin-1-yl-ethoxy)-phenyl)] moiety at the 6-site of the pyrazolo [1, 5-a]pyrimidine scaffold was replaced by dimethylamino, diethylin, 3-methyl-piperidyl, 2-methyl-pyrrolyl or 1-azetidinyl. We found that these compounds inhibited heat stress-induced HSP70 expression with different efficacies (Figure 4, Table 1). When R1 was not changed and R2 was substituted with other moieties, the derived compounds 1#, 2#, and 4# inhibited heat-induced HSP70 expression with an IC50 comparable to that of dorsomorphin, and compounds 3# and 5# had a slightly higher IC50 than that of dorsomorphin. When R1 was substituted with 4-isoquinolyl and R2 was simultaneously replaced with 1-azetidinyl, the derived compound (20#) also had a similar inhibitory effect as dorsomorphin on heat-induced HSP70 expression. Compounds derived from other substitutions of the R1 and R2 moieties all had lower inhibitory activities than that of dorsomorphin (Table 1). These results indicate that the 4-pyridyl at the 3-site of the pyrazolo [1, 5-a] pyrimidine ring is critical for the potent inhibitory activity of dorsomorphin on heat shock response; the combination of 4-isoquinolyl (R1) and 1-azetidinyl (R2) in the pyrazolo [1, 5-a] pyrimidine scaffold also contributes to the inhibitory effect on heat shock response. We further found that in addition to suppressing heat-induced HSP70 mRNA expression, compounds 1#, 2#, 4#, and 20# inhibited heat-induced HSP70 protein expression (Figure 4A and 4B) and HSF1 nuclear translocation (Figure 4C). All these compounds significantly reduced cancer cell viability and induced apoptotic cell death (Figure 4D and 4E).
Effect of DM and its derivatives on heat stress-induced HSP70 expression
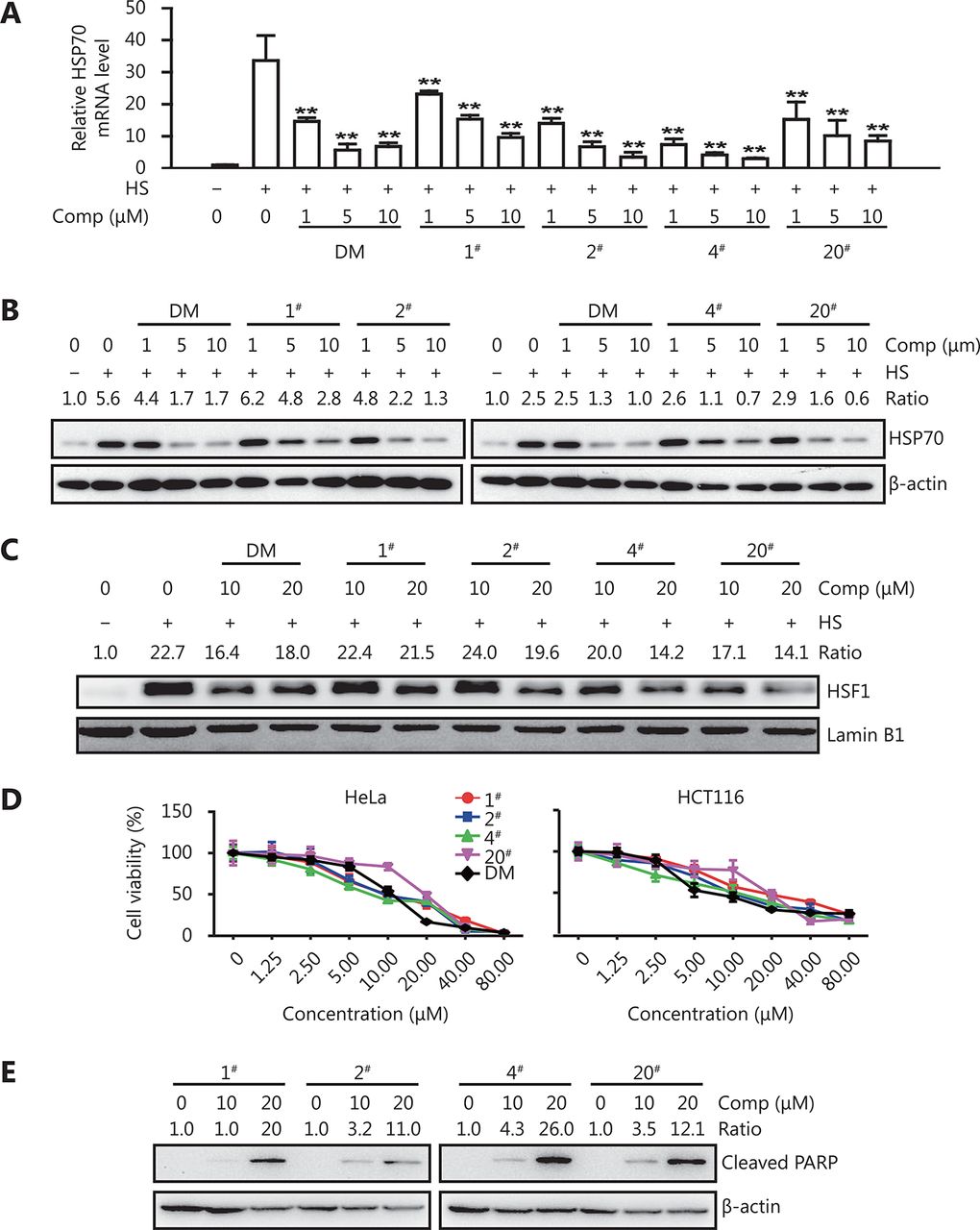
DM derivatives inhibit heat shock response and induce cancer cell apoptosis. (A-C) HeLa cells were treated with different concentrations of DM or its derivatives (compounds 1#, 2#, 4# and 20#) for 30 min and then exposed to 42°C (heat stress, HS) for 1 h. HSP70 mRNA (A) and nuclear HSF1 (C) levels were examined by qRT-PCR and Western blot, respectively; HSP70 protein levels were examined by Western blot after recovery at 37°C for 5 h (B). (D-E) HeLa or HCT116 cells were incubated with different concentrations of DM or its derivatives for 24 h and examined for cell viability with CCK-8 (D) or cleaved PARP by Western blot (E, HeLa cells). Data are shown as mean ± SD, n = 3. *P < 0.05, **P < 0.01 vs. heat-exposed cells. The images in B, C and E are representative results of three independent experiments.
Dorsomorphin sensitizes cancer cells to both HSP90 and proteasome inhibitors and suppresses HSP upregulation by these inhibitors
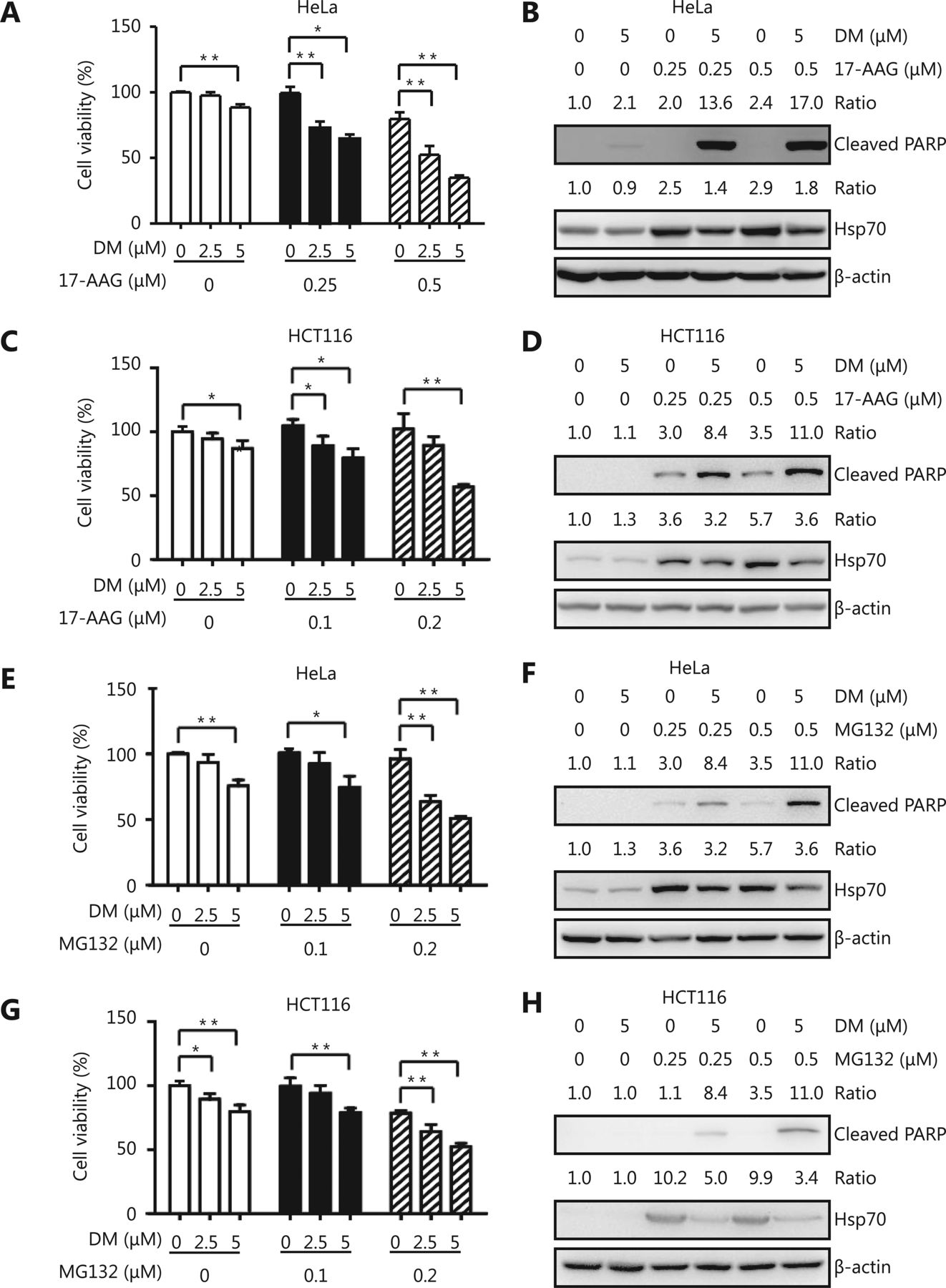
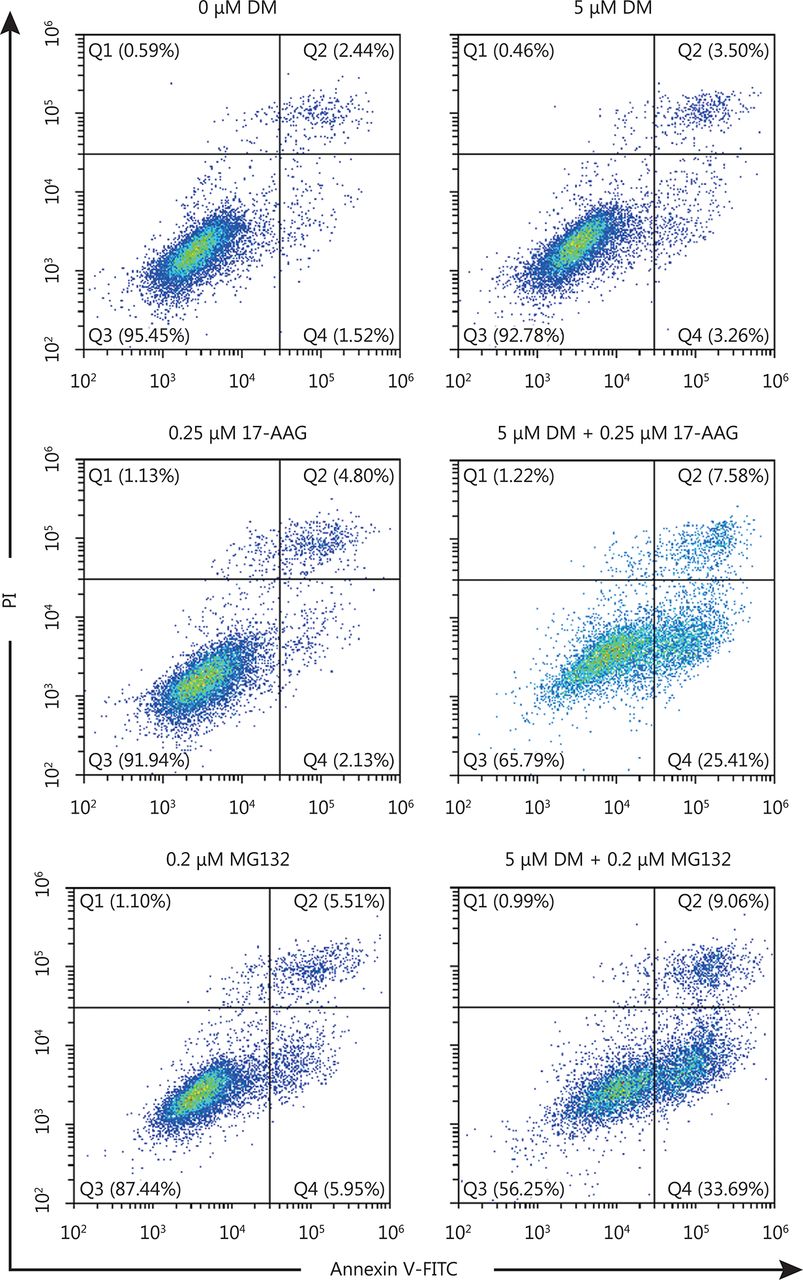
HSP90 inhibitors and proteasome inhibitors are anti-cancer agents in clinical trials. A problem with most of these compounds is that they potently activate HSF1, which leads to HSP expression and protects cancer cells from apoptosis. Because dorsomorphin could inhibit HSF1 activation by reducing its nuclear translocation, it may reduce the side effect of HSP90 and proteasome inhibitors. We found that while treatment with low concentrations of dorsomorphin or the HSP90 inhibitor 17-AAG had no significant effect on the viability of HeLa and HCT116 cells, a combination of these two compounds synergistically reduced the viability of these cells (Figure 5A, 5C) and induced apoptotic cell death (Figure 5B, 5D and Supplementary Figure S2). Furthermore, dorsomorphin inhibited 17-AAG-induced HSP70 expression in these cancer cells (Figure 5B and 5D). Similarly, dorsomorphin enhanced the inhibition of cell viability (Figure 5E and 5G) and induction of apoptosis of cancer cells by the proteasome inhibitor MG132, and suppressed MG132-induced HSP70 expression (Figure 5F and 5H and Supplementary Figure S2). Altogether, our results demonstrate that dorsomorphin can enhance the pro-apoptotic effect of both HSP90 and proteasome inhibitors on cancer cells and reduce HSP70 upregulation induced by these inhibitors.
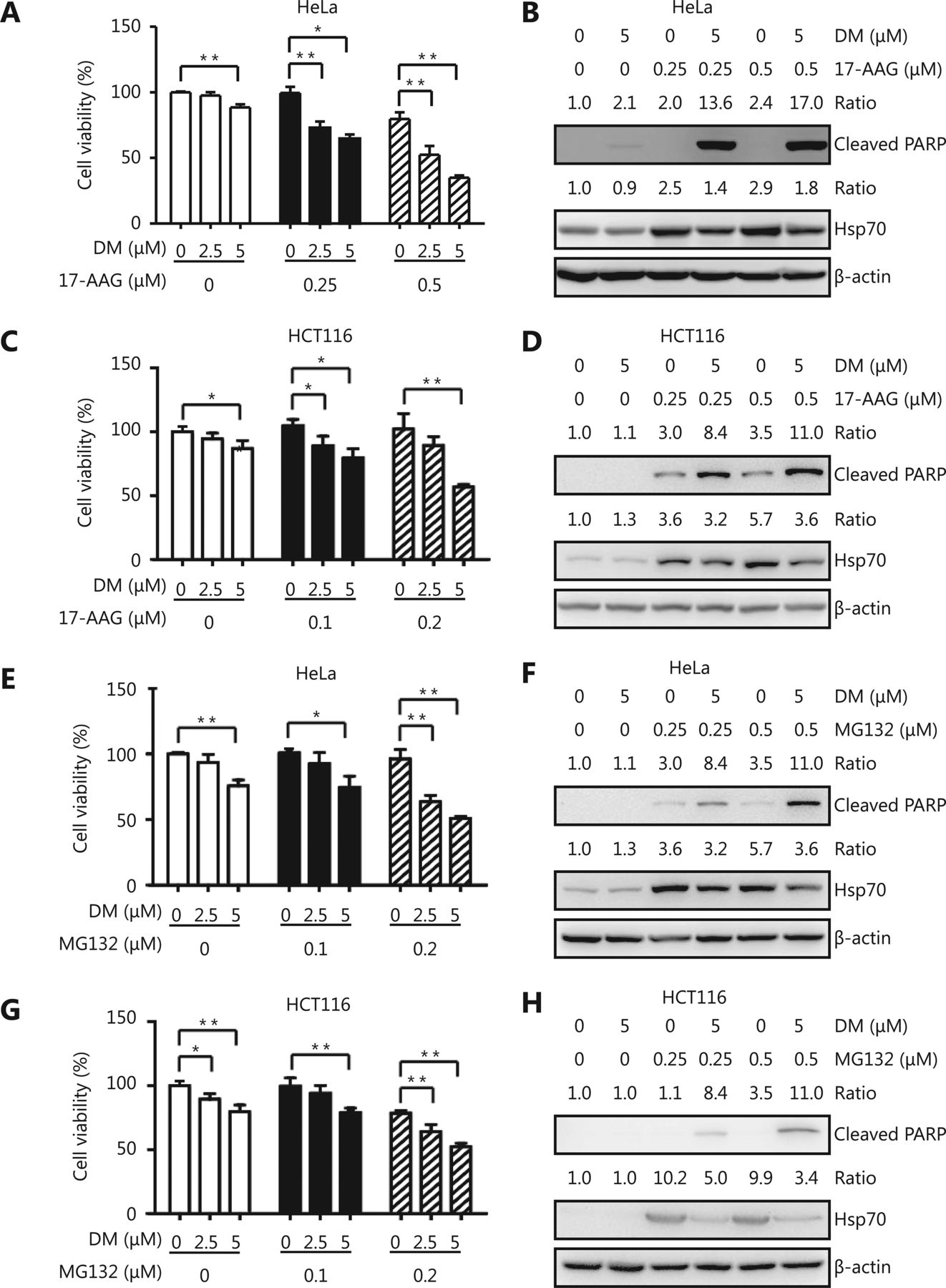
DM induces cancer cell apoptosis synergistically with an HSP90 inhibitor or a proteasome inhibitor and suppresses HSP upregulation by these inhibitors. Cancer cells were incubated with different concentrations of DM and 17-AAG or MG132 for 24 h, cell viability was examined with CCK-8 (A, C, E, G), and cleaved PARP and HSP70 were examined by Western blot (B, D, F, H). Cell viability was shown as means ± SD, n = 3-5. *P < 0.05, **P < 0.01, the images in B, D, F and H are representative results of three independent experiments.
Dorsomorphin suppresses tumor growth synergistically with 17-AAG and inhibits 17-AAG-induced HSP expression in tumor tissues
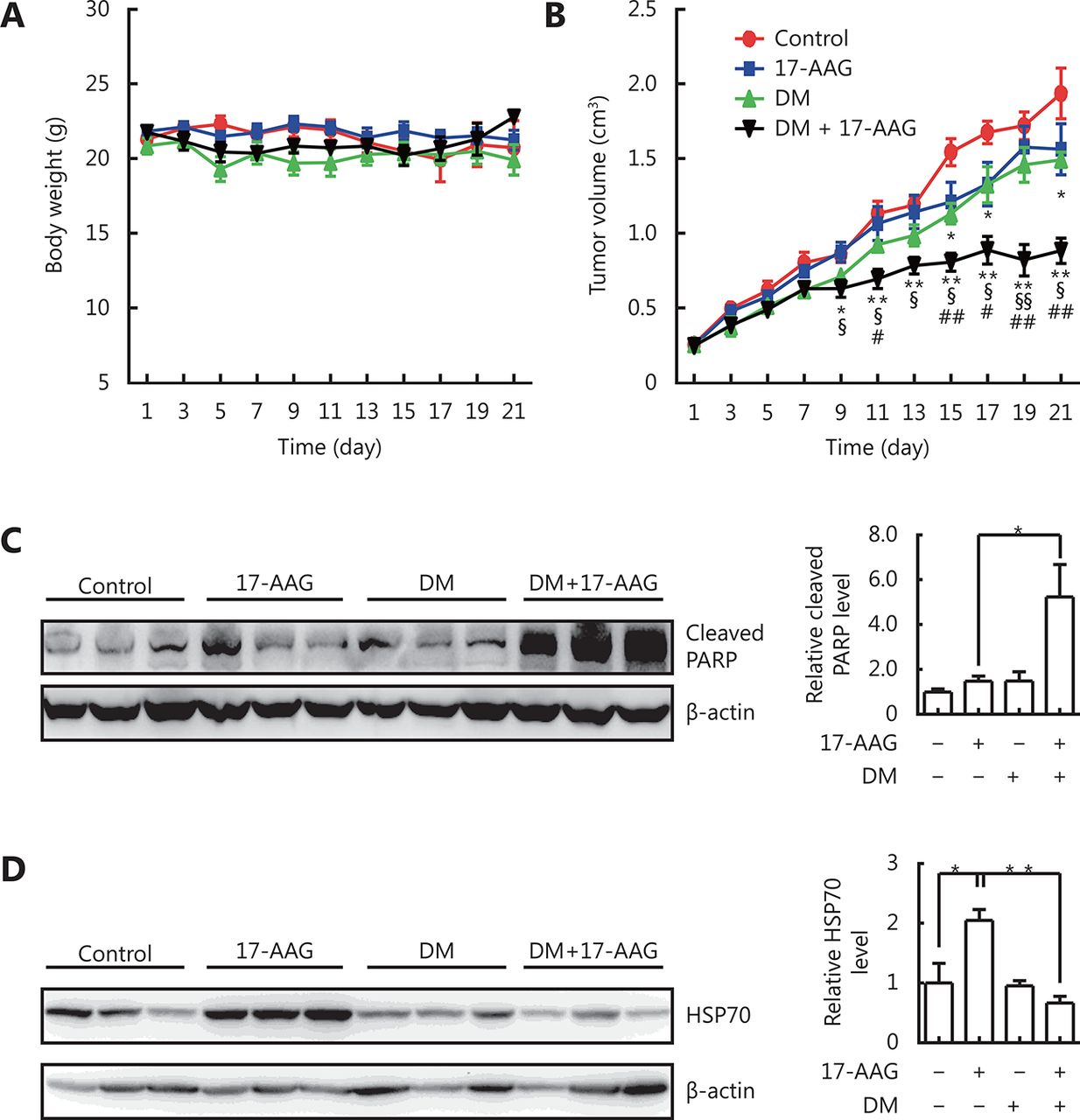
We further investigated whether dorsomorphin could enhance the anti-tumor effect of 17-AAG and inhibit its induction of HSP70 expression in tumor tissues by using tumor-bearing nude mice. Mice bearing HCT116 tumors received vehicles, dorsomorphin (10 mg/kg weight, twice per week), or dorsomorphin combined with 17-AAG (50 mg/kg body weight, thrice per week) for 3 weeks. Treatment of mice with these compounds had no significant effect on body weight (Figure 6A). While low-dose 17-AAG treatment had no significant effect on tumor growth and low-dose dorsomorphin treatment slightly inhibited tumor growth, a combination of these two compounds significantly inhibited tumor growth (Figure 6B) and increased the cleaved PARP levels in tumor tissues (Figure 6C). Furthermore, we found that 17-AAG markedly induced HSP70 expression in tumor tissues, and the combination of dorsomorphin with 17-AAG could suppress HSP70 expression induced by 17-AAG (Figure 6D). These results demonstrate that dorsomorphin can enhance the anti-tumor effect of 17-AAG and inhibit 17-AAG-induced HSP70 expression in tumor tissues.
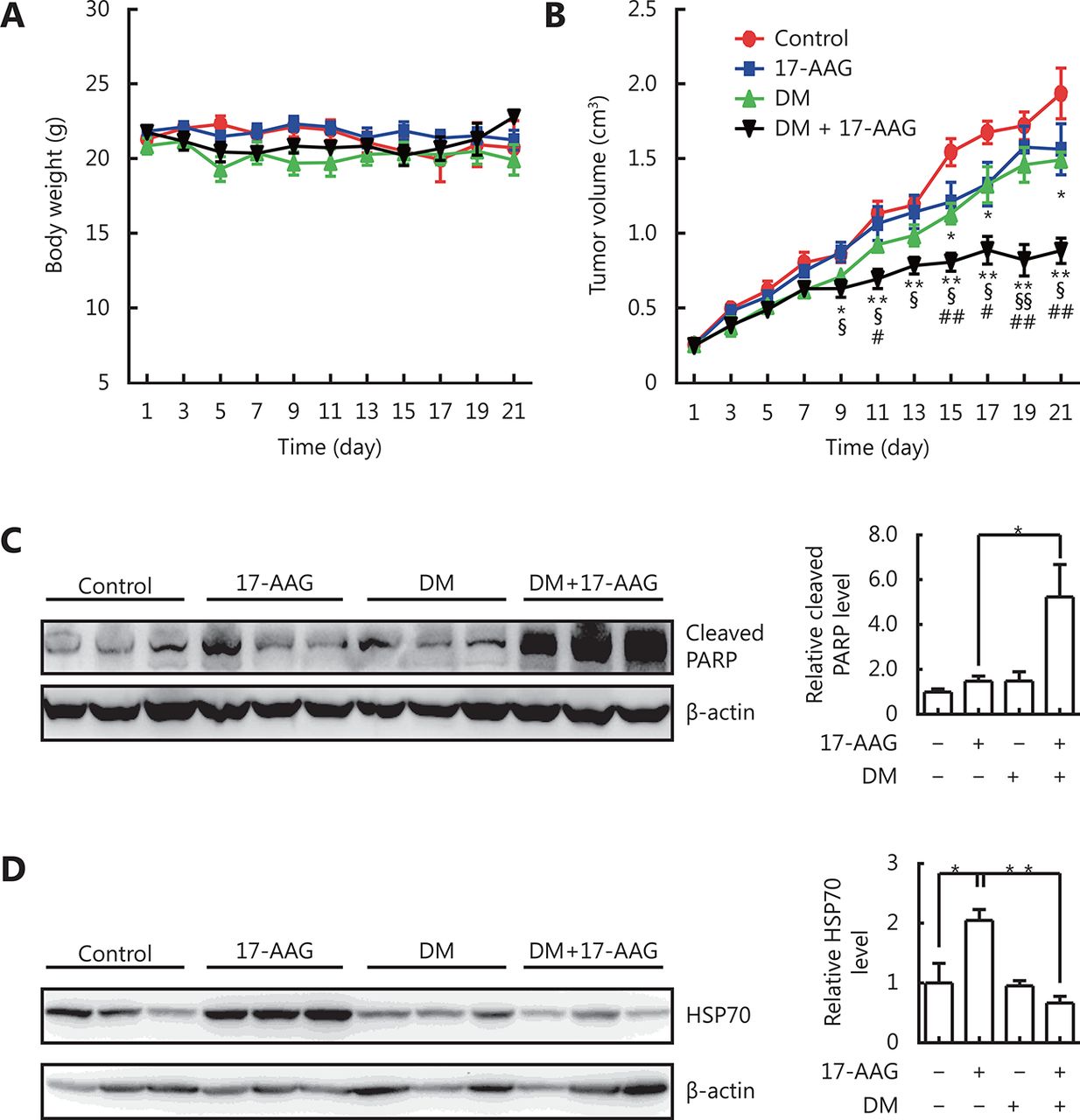
DM enhanced anti-tumor effect of 17-AAG and inhibited 17-AAG-induced HSP70 expression in tumor tissues. HCT-116 cells were implanted subcutaneously into the right flank of nude mice. When the tumor grew to approximately 250 mm3, the mice were divided into four groups: control, 17-AAG, DM, and DM + 17-AAG (n = 6-9 per group). DM·2HCl (10 mg/kg body weight) was injected into the tumor twice a week, and 17-AAG (50 mg/kg body weight) was injected intraperitoneally three times a week. The control mice were administered vehicles. The body weight (A) and tumor size (B) were measured every other day. The mice were sacrificed on the 21st day and examined for cleaved PARP (C) and HSP70 expression in the tumor tissue by Western blot (D). Data were shown as means ± SE. *P < 0.05, ** P < 0.01, comparing the control group with the DM group or (17-AAG + DM) group (B); §P < 0.05, §§P < 0.01, comparison between the (17-AAG + DM) group and the 17-AAG group; #P < 0.05, ##P < 0.01, comparison between the (17-AAG + DM) group and the DM group. Images in C and D are representative results of three independent experiments.
Discussion
In the present study, we found that dorsomorphin inhibited constitutive and multiple stimuli-induced HSP expression and induced cancer cell apoptosis. Mechanistic studies revealed that dorsomorphin induced cancer cell apoptosis through inhibiting HSF1-mediated HSP expression. Structure-activity analysis showed that the 4-pyridyl at the 3-site in the pyrazolo [1, 5-a] pyrimidine scaffold of dorsomorphin was critical for its potent inhibition of HSF1 nuclear translocation and HSP expression. Both in vitro and in vivo studies demonstrated that dorsomorphin not only enhanced the anti-tumor effect of both HSP90 and proteasome inhibitors but also repressed HSP70 expression induced by these inhibitors.
HSF1 is a major transcription factor regulating HSP expression in heat shock response induced by physiological and environmental stimuli33. HSPs aid in the refolding of misfolded peptides and restrain protein aggregation, which maintains protein homeostasis and protects cells and organisms against severe stress insults. However, HSF1 expression is elevated or activated in various types of malignant tumors and plays critical roles in tumorigenesis and progression through both HSP-dependent and -independent pathways. HSF1 is an attractive target for cancer therapy. Several compounds have been reported to induce cancer cell apoptosis through inhibiting HSF1-mediated HSP expression, including promoting HSF1 degradation by reducing HSF1 stability, inhibiting HSF1 phosphorylation or HSF1 binding with a heat shock element in the promoter region of HSP genes34-36, inhibiting HSF1 transcription activity by impairing the recruitment of positive transcription elongation factor b to the HSP promoter37 or preventing HSF1 binding to activating transcription factor38, and inhibiting the transactivation function of HSF139. However, no compound has been reported to suppress HSP expression through inhibiting HSF1 nuclear translocation.
In this study, we found that dorsomorphin not only inhibited HSP expression induced by heat stress, HSP90/proteasome inhibitors, and cadmium (Figure 1A–1C), but also suppressed constitutive HSP70 expression in cancer cells (Figure 1D and 1E). Further studies showed that dorsomorphin had no influence on the expression of HSF1, but markedly repressed HSF1 nuclear translocation under heat stress. In support of the inhibitory effect of dorsomorphin on HSF1 nuclear translocation, we found that dorsomorphin suppressed heat stress-induced formation of nuclear HSF1 granules (Figure 2C), a nuclear compartment for the temporal regulation and spatial organization of HSF1 activity40,41. Besides inhibition of HSF1 nuclear translocation in heat-stressed cells, dorsomorphin also reduced nuclear HSF1 level under resting conditions. These results indicate that dorsomorphin inhibits HSP expression through suppressing HSF1 nuclear accumulation. Overexpression of constitutively active HSF1 in cancer cells increased nuclear HSF1 level and reversed dorsomorphin-induced HSP70 inhibition and cancer cell apoptosis, indicating that dorsomorphin induces cancer cell apoptosis through inhibiting HSF1 activation and HSP70 expression. HSF1 activation is a complex process, the mechanisms involved in HSF1 nuclear translocation have not been fully elucidated. It has been reported that phosphorylation of HSF1 on Ser419 by polo-like kinase 1 and on Ser320 by protein kinase A contributes to HSF1 nuclear translocation during heat stress30, 31. We found that dorsomorphin inhibited heat stress-induced phosphorylation of HSF1 on Ser320 (Figure 2D), indicating that dorsomorphin induces HSF1 nuclear translocation through phosphorylating HSF1 at Ser320. There is no specific antibody against Ser419 of HSF1 currently available, and further investigation is needed to determine whether dorsomorphin suppresses HSF1 nuclear accumulation through inhibiting HSF1 phosphorylation at Ser419. Phosphorylation of HSF1 at Ser303 and Ser307 has been reported to be involved in 14-3-3ε binding to HSF1, which facilitates HSF1 nuclear export3,32. We found that dorsomorphin had no influence on the phosphorylation of HSF1 at Ser320, Ser 303, and Ser 307 in cancer cells under resting conditions. The mechanisms underlying the reduction of nuclear HSF1 by dorsomorphin in cancer cells under resting conditions remain unknown.
Dorsomorphin is commonly used as an AMPK inhibitor. We found that knockdown of AMPKα did not affect the inhibition of HSP70 expression by dorsomorphin. This outcome indicates that dorsomorphin inhibits HSP70 expression independent of AMPK. Our previous studies showed that heat stress inhibited AMPK activity. Heat stress-induced HSP70 expression was inhibited by AICAR, an AMPK activator. Further, knockdown of AMPKα by RNA interference reversed the inhibitory effect of AICAR on heat stress-induced HSP70 expression, indicating that the inhibition of AMPK by heat shock promotes HSP expression28. These results suggest that the inhibition of HSP expression by dorsomorphin is not mediated through AMPK.
We studied the structure-activity of dorsomorphin and found that compounds 1#, 2#, 4#, and 20# were as effective as dorsomorphin in inhibiting heat stress-induced HSF1 nuclear translocation and HSP70 expression, repressing cancer cell viability, and inducing cancer cell apoptosis. The 4-pyridyl at the 3-site in the pyrazolo [1, 5-a]pyrimidine scaffold of dorsomorphin played an important role in inhibiting HSF1 activation and inducing cancer cell apoptosis. These findings are helpful for designing new HSF1 inhibitors.
As HSF1 contributes to tumorigenesis through both HSP-dependent and -independent mechanisms, inhibition of HSF1 by dorsomorphin and its derivatives may suppress not only HSP expression but also the other pathways activated by HSF1 in cancer cells. Through in vitro and in vivo studies, we demonstrated that the combination of dorsomorphin and an inhibitor of HSP90 or proteasome not only synergistically enhanced the inhibitory effect of these two drugs on cancer cell viability, promoted cancer cell apoptosis, and inhibited tumor growth, but also inhibited HSP70 expression induced by these two drugs in cancer cells and tumor tissues. Therefore, in addition to inducing cancer cell apoptosis through inhibiting HSF1 and sensitizing cancer cells to HSP90 and proteasome inhibitors, dorsomorphin and its derivatives may prevent cancer cell resistance to these inhibitors through blocking the activation of HSF1.
In summary, we identified dorsomorphin as an HSF1 inhibitor. By inhibiting HSF1 nuclear accumulation, dorsomorphin inhibits the expression of HSP70 and other HSPs, promotes cancer cell apoptosis, enhances the anti-tumor effect of HSP90 and proteasome inhibitors, and may prevent tumor resistance to these two types of drugs by inhibiting the induction of HSPs. Hence, dorsomorphin and its derivatives may serve as potential therapeutic agents for cancer treatment.
Acknowledgments
This study was supported by grants from the National Key Research and Development Program of China (Grant No. 2017YFC1601702) and China Postdoctoral Science Foundation (Grant No. 2011M500825). We thank Professor Sandy Westerheide at the University of South Florida for providing the constitutively active HSF1-expressing plasmid pcDNA3.1-HSF1.
Conflict of interest statement
No potential conflicts of interest are disclosed.
Supplementary material
The primer sequences used for qRT-PCR:
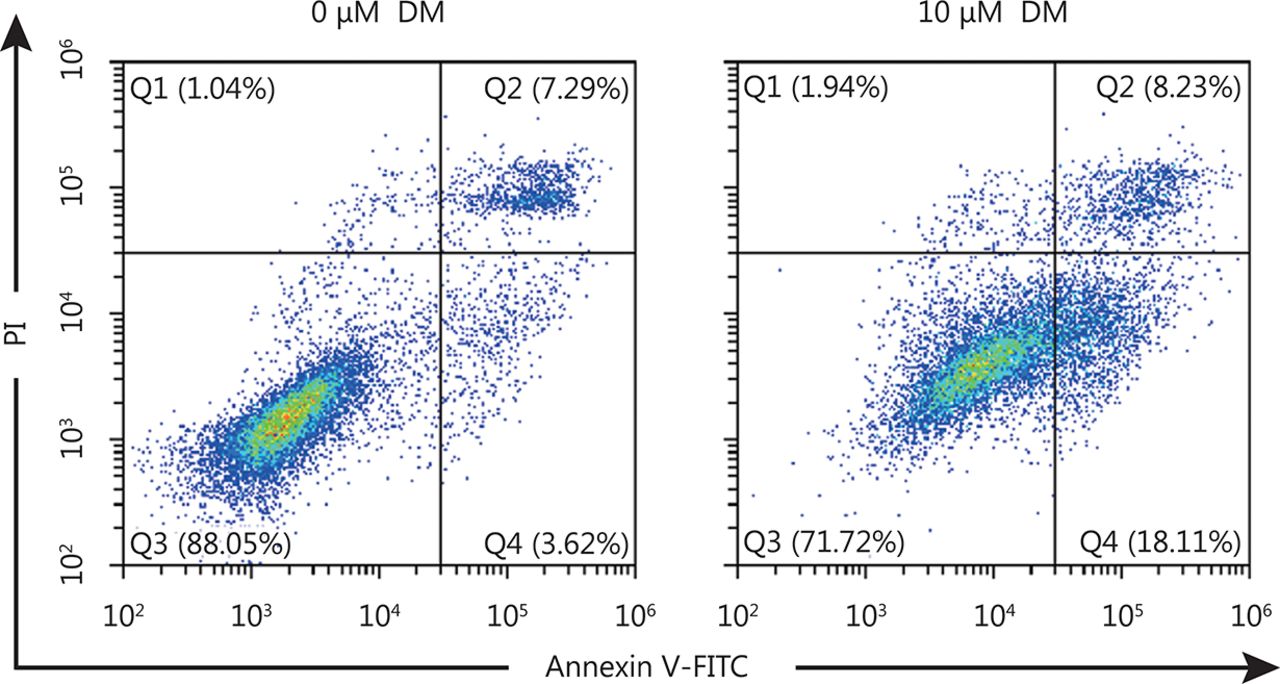
HeLa cells were incubated with 10 μM DM for 24 h, cells were stained with Annxin V-FITC and PI, examined with a flow cytometry. Q2 represents late stage apoptotic cells and Q4 represents early stage apoptotic cells.
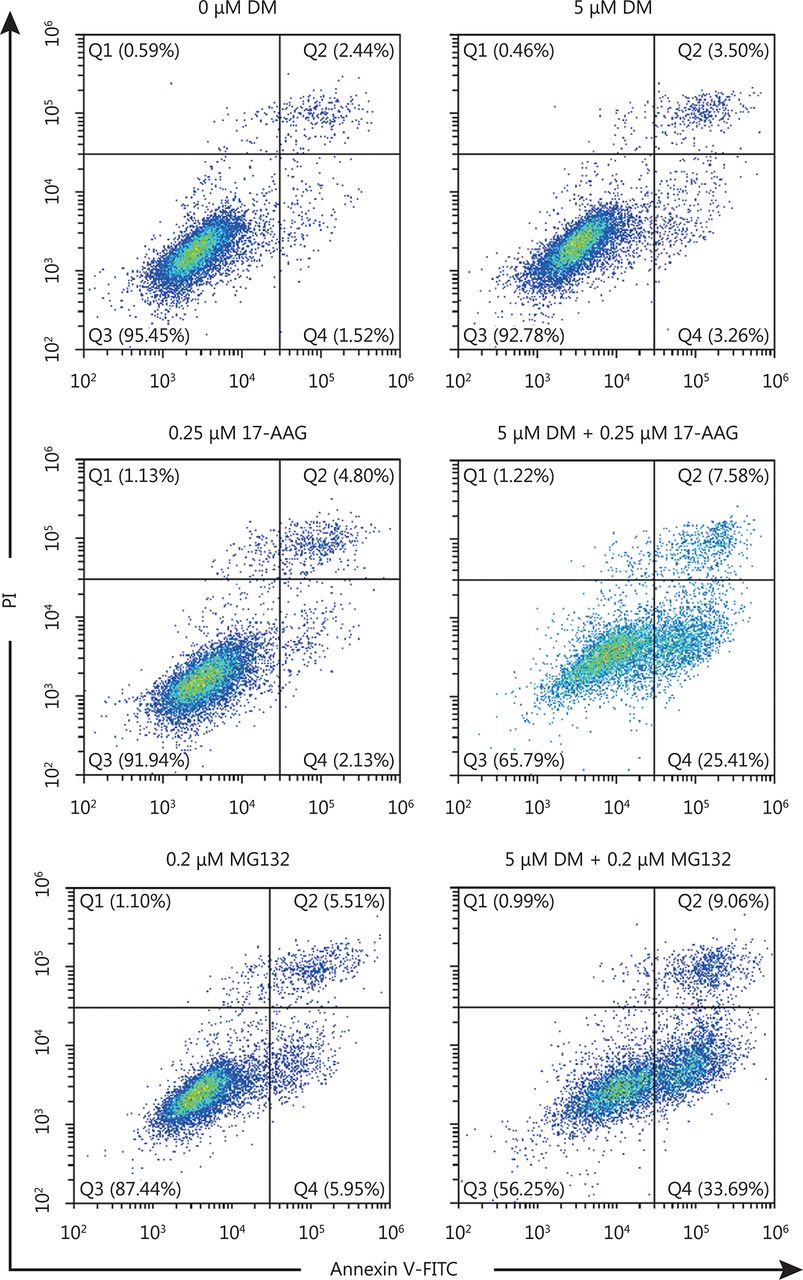
HeLa cells were incubated with different concentrations of DM and 17-AAG or MG132 for 24 h, cells were stained with Annxin V-FITC and PI, examined with a flow cytometry. Q2 represents late stage apoptotic cells and Q4 represents early stage apoptotic cells.
- Received August 6, 2018.
- Accepted January 2, 2019.
- Copyright: © 2019, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
In this issue






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.