

Abstract
OBJECTIVE To determine the effect of short interference RNA (siRNA) against STAT3 induced inhibition of STAT3 gene expression and on the growth and apoptosis of Lewis lung cancer cells.
METHODS pSilencer 2.1 -U6 STAT3 siRNA against STAT3-mRNA was synthesized. Lewis lung cancer cells were divided Into 3 groups: vehicle, plasmid, and STAT3 siRNA in which the cells were treated with RPMI-1640 culture media, or transfected with pSilencer empty vector, or pSilencer STAT3 siRNA. Semiquantitative RT-PCR and Western blot analysis of STAT3 gene expression in the cells was performed 72 h after transfection. MTT assay for cell proliferation, flow cytometry and DNA laddering electrophoresis were used for determination of cell proliferation and apoptosis.
RESULTS STAT3 was markedly expressed at both the mRNA and protein levels in the cells treated with RPMI-1640 media or transfected with the plasmid vector, whereas STAT3 expression was significantly reduced in cells treated with STAT3 siRNA. These findings suggest that STAT3 siRNA effectively inhibited STAT3 expression. Transfection of the cells with STAT3 siRNA resulted in significant cellular growth inhibition and enhanced apoptosis.
CONCLUSION Transfection of Lewis lung cancer cells with synthetic STAT3 siRNA resulted in effective Inhibition of STAT3 gene expression at both protein and mRNA levels, leading to Induced apoptosis and growth suppression.
keywords
The morbidity from lung cancer, one of the most common primary malignant carcinomas in the world, has increased annually. Furthermore, to date, the therapeutic efficacy for this malignancy has not improved as manifested by current high mortality and poor long-term survival rates. Proteins, which are signal transducers and activators of transcription (STAT), perform the dual functions of signal transduction and activation of transcription.[1] An increasing number of studies have suggested that STATs have the potential to be novel molecular targets to treat lung cancers.[2-4]
STATs are latent cytoplasmic transcription factors that function as intracellular effectors of cytokine and growth factor signaling pathways. [2] They were originally described in the context of regulating cell signaling, contributing to such diverse process as differentiation, proliferation, and apoptosis. It has been well documented that activation of all of the STAT proteins is caused by phosphorylation of a single tyrosine residue that leads to dimerization via an intermolecular SH2 phosphotyrosine interaction.[5, 6] The dimerized STATs translocate to the nucleus where they regulate gene expression by binding directly to high affinity DNA binding sites or by associating with other transcription factors.[7] In normal and/or benign cells, the signaling by STAT3 is under tight regulation so that the signal is transient. How-ever, aberrant signaling by STATs has been reported in many types of malignancies, such as myeloma, head and neck, breast, and prostate cancers.[7-9] Amongst the Stat family, STAT3 plays a key role in promoting pro-liferartion, differention, and anti-apoptosis.[2, 3, 10]
To date, constitutive activation of STAT3 has been detected in a number of tumour-derived cell lines, as well as in a wide variety of human malignancies.[3, 11-19] We have recently reported that the expression of the STAT3 gene is significantly increased in human laryngeal cancer cell lines (Hep2) and prostate cancer cells at both mRNA and protein levels.[2] Seki et al.[20] have found a significantly increased expression of the STAT3 gene in human lung cancer tissues. Thus, it is plausible that constitutive activation of STAT3 represents an important role in the growth and survival of cancer cells.
It is known that dysregulation of apoptosis contributes to cancers and many diseases. Studies have shown that over active STAT3 promotes uncontrolled growth and survival through aberrant expression of downstream targeted genes, such as cyclin Dl, cyclin D2, c-Myc, p53, Bcl-xL, Bcl-2, Mcl-1, and survivin; [21-23] these genes influence cell cycle progression or in-hibit apoptosis. The up-regulation of these genes in cancers has been well-documented, and it has been demonstrated that STAT3 can directly regulate expression of several survival genes.[5-7] The anti-apoptotic genes encoding Bcl-xL, Mcl-1, and survivin proteins are STAT3 target genes.[5-7] It has been reported in studies with breast cancer cells that activation of STAT3 signaling induces enhanced expression of Mcl-1 and survivin genes with anti-apoptotic activity, whereas disruption of STAT3 signaling results in a dramatic reduction in the expression of Mcl-1 and survivin and induction of apoptosis.[16, 24-26] Our recent studies along with others have demonstrated simultaneously increased STAT3 gene expression and decreased expression of Bcl-2 or Bcl-xL genes in human laryngeal cancer cell lines (Hep2), prostate cancer cells, and astrocytoma cells at both mRNA and protein levels.[2, 3, 10, 27] Likewise, the knockdown of STAT3 expression by RNA interference (RNAi), significantly increased expression of Bcl-2 or Bcl-xL genes, suggesting induction of apoptosis in human Hep2 cells, prostate cancer cells, and astrocytoma cells.[2, 3, 10, 27]
There is evidence to suggest that malignant cells having constant activation of STAT3 become STAT3-dependent for survival, and that deactivation or down-regulation of STAT3 results in apoptosis.[5,7] Studies have shown that using a variety of approaches, such as tyrosine kinase inhibitors, antisense oligonucleotides, decoy oligonucleotides, and dominant-negative STAT3 proteins can inhibit STAT3 expression in cancer cells to suppress proliferation and induce apoptosis. [28-31] In human head and neck squamous carcinoma cells, blocking of STAT3 signaling by decoy oligonucleotides or antisense oligonucleotides inhibits transforming growth factor effects and suppresses oncogenic growth of these cells.[7, 8] STAT3β is a naturally occurring dominant negative STAT3 variant that is identical to STAT3 except for the absence of the transactivation domain.[9] Blockade of STAT3 signaling by STAT3β in human myeloma cells and breast cancer cells in vivo down-regulates IL-6-induced expression of the Bcl-xL antiapoptotic gene and induces apoptosis, [32] thereby suggesting that targeting STAT3 by STAT3β may enhance in vivo antitumor responses.
The use of RNA interference (RNAi) represents a novel alternative to gene inhibition that may be capable of inhibiting STAT3 expression in cancer cells leading to suppression of proliferation and induction of apoptosis. [28-30] RNAi is triggered by introducing long double-stranded RNA (dsRNA) molecules into cells where the dsRNAs are cleaved by an endonuclease named dicer into 21- to 23-nt RNAs referred to as short interference RNAs (siRNAs).[33] The siRNA molecules that serve as a guide for sequence-specific degradation of homologous mRNAs have been used for functional analysis of genes in many species.[34] siRNA targeting STAT3 has been successfully used for treating numerous cancers. [2, 3, 27] However, to our knowledge, no reports have been published to date concerning the effect of siRNA against the STAT3 gene in lung cancers cells.[20] In our studies we have used Lewis lung cancer cells, which have shown to be of value for the study of lung cancer.
The objectives of the present study were (1) to determine if STAT3 siRNA can inhibit the expression of STAT3 gene in Lewis lung cancer cells and (2) to investigate the effect of STAT3 siRNA on the growth and apoptosis in these cells.
MATERIALS AND METHODS
Experimental protocols
pSilencer 2.1-U6 STAT3 siRNA against STAT3-mRNA was synthesized. Lewis lung cancer cells were divided into three groups: vehicle, plasmid, and STAT3 siRNA where the cells were treated with RPMI-1640 culture media, or transfected with pSilencer empty vector, or pSilencer STAT3 siRNA. Semiquantitative RT-PCR and Western blot analysis of STAT3 gene expression was performed at 72 h after transfection. MTT [3-(4,5 -dimethylthiazol-2-yl)-2,5 -diphenyltetra-zolimium] assay for cell proliferation, flow cytometry and DNA laddering electrophoresis were used for determination of cell proliferation and apoptosis, respectively.
Construction of plasmids and siRNA expression
A double stranded siRNA oligonucleotide against STAT3 was constructed using our previously described method.[2] The pSilencer2.1-U6 (Ambion Inc. Austin, TX) was used for DNA vector-based siRNA synthesis under the control of the U6 promoter in vivo. In brief, first, the double stranded DNA template encoding siRNA oligonucleotides (GeneBank: access numbers for the human STAT3: NM003150) was synthesized that contained a sense strand of 19 nucleotide sequences with homology to the mouse STAT3 gene sequences followed by a short space (TTCAAGAGA), the reverse complement of the sense strand, and five thymidines as a RNA polymerase III transcriptional stop signal. The sequences were forward, 5’-GCA GCA GCT GAA CAA CAT GTT CAA GAG ACA TGT TGT TCA GCT GCT GCT TTT TT-3’ and reverse, 5’-AAT TAA AAA AGC AGC AGC TGA ACA ACA TGT CTC TTG AAC ATG TTG TTC AGC TGC TGC GGC C-3’ (located on SH2 domain). The oligonucleotides were placed in a buffer (potassium acteate 100 mmol/L, 30 mmol/L HEPES-KOH pH 7.4, and magnesium acetate 2 mmol/L) and the mixture incubated at 90°C for 3 min and then at 37°C for 1 h. The double stranded oligos were cloned into the Apal-EcoR I sites of the pSilencer 2.1-U6 vector where short hairpin RNAs (shRNA) were expressed under the control of the U6 promoter. A vector (Ambion Inc.) that expresses a hairpin siRNA with limited homology to any known sequences in the human, mouse, and rat genomes was used as a negative control. The negative control siRNA had no toxicity on cells tested by trypan blue staining 48 h after transfection, and also no effect on the expression of the mRNA levels of “housekeeping” genes, including 28S rRNA, GAPDH, and Cyclophilin (Ambion).
Cell culture and Iransfection
Cell culture and transfection were conducted using our methods previously described.[2] The Lewis lung cancer cell line was obtained as a gift from the Department of Pathology, School of Basic Medicine, Jilin University. The cells were cultured in RPMI 1640 (Invitrogen, Inc, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and penicillin (100 kU/L) and streptomycin (100 mg/L) at 37°C in a humidified incubator with 5% C02. For cell transfection, Sofast gene transfection reagent (Sunma Biotechnology Ltd) was used for transfecting the plasmids under the manufacturer instructions. In brief, pEGFP was cotransfected with pSilencer2.1-U6-siRNA-STAT3 or pSilencer empty vector at a ratio of 1:20 to mark the positive transfected cells, respectively. The cells were cultured for 5~20 h and then transferred to fresh medium with 10% FBS and lysed at 24~72 h after transfection. The cells were cultured for 72 h after transfection and than lysed.
Cell proliferation
For the cell proliferation assay, cells were transfected with siRNA-STAT3 or pSilencer negative vector incubated for 72 h in 96-well plates. Cellular proliferation was determined by the MTT assay using a hemocytometer to count cells in Absorbance at 570 nm (A570) was determined on a multiwell plate reader. Cell growth inhibition rate was calculated according to the following formula:
Growth inhibition rate (%)=[(A570c-A570e )/A570c] ⨯ 100 %.
A570c: A570 in control group; A570e: A570 in experimental group.
Flow cytometric analysis of apoptosis
For flow cytometric analysis of apoptosis, the cells were transfected with STAT3 siRNA or a pSilencer empty vector for 72 h. The cells were collected and washed with cold PBS containing 4 mmol/L EDTA. Cells were fixed in 70% cold ethanol, collected by centrifugation, and washed once again with PBS containing 4 mmol/L EDTA. Thereafter, the cells were resuspended in PBS containing 4 mmol/L EDTA, 20 mL/L of propidium iodide (Sigma Chemical), 0.2% Triton X-100, 40 mg/L RNase A, and incubated for at least 30 min at 4°C. Finally, the cells were analyzed by flow cytometry (FACScan, Becton Dickinson, Franklin Lakes, NJ, USA), using Cell Quest software.
DNA laddering electrophoresis
DNA fragmentation was measured by gel electrophoresis. Briefly, to detect intemucleosomal cleavage of the DNA, the presence of low-molecular-weight DNA fragments was determined in the cells cultured for 72 h in the presence of STAT3 siRNA or a pSilencer empty vector. The cells were plated at a density of 5⨯106 cells/plate and a concentration of lxlO6 cells/ mL and then were pelleted and resuspended in lysis buffer (50 mmol/L Tris HCl, 10 mM EDTA, 0.5% SDS). Subsequently, the cell lysates were treated with 20 mg/mL proteinase K and incubated at 55 °C for 1 h followed by addition of 0.5 mg/mL RNAse A and then heated to 70 °C for 5 min. DNA was precipitated with isopropanol and mixed with 12 ¼l of loading dye (10 mM EDTA, pH 8.0, 40 % sucrose, 0.25 % bromophenol blue). The samples were subjected to electrophoresis on a 2% agarose gel containing 0.5 (¼g/ml ethidium bromide. DNA in the gels was visualized under ultraviolet light. The gel images were captured using an Alpha Innotech digital camera equipped with a transilluminator and Alpha Ease 5.5 software (Alpha Innotech Corporation, San Leandro, CA).
Western blot analysis
Western blotting analysis of STAT3 expression in the cells was accomplished using our previously described method.[2] Total protein was extracted from the harvested sample cells with protein lysis buffer (5 mol/L EDTA, 300 mmol/L NaCl, 0.1% Igepal, 0.5 mmol/L NaF, 0.5 mmol/L Na3VO4, 0.5 mmol/L PMSF, and antiprotease mixture) using sonication. The lysates were centrifiiged at 15,000 g for 30 min. Determination of protein concentrations of the supernatants was performed by the Bradford procedure (Bio-Rad Laboratory, Hercules, CA). For STAT3 and β-actin analysis using antibodies against STAT3 and β-actin (Santa Cruz Biotech, Santa Cruz, CA), the supernatant with 50 ¼g total protein was separated by electrophoresis on 10% SDS-Polyacrylamide gels and transferred onto PVDF membranes (Milipore, Bedford, MA) and blocked with 5% nonfat dry milk in PBS with 0.1% Tween 20. Blots were incubated with specific rabbit antibodies against STAT3 and anti-β-actin antibody and washed with TBST and subjected to corresponding HRP-conjugated secondary antibodies as indicated. Blots were washed again with TBST and visualized by an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Uppsala, Sweden).
RT-PCR
Semi-quantitation of specific mRNA by RT-PCR was performed as described previously. Total RNA was extracted from the collected sample cells with Trizol (Invitrogen) following the manufacturer’s instructions For RT-PCR analysis, 3 µg of total RNA was subjected to reverse transcription using a RT-PCR kit (Promega, Madison, WI). For amplification of STAT3 mRNA, the primer pairs 5’-TTG CCA GTT GTG GTG ATC-3’ and 5’-AGA ACC CAG AAG GAG AAG C-3’ were used with the following conditions: 94 °C for 5 min, then 30 cycles of 94 °C for 30 s, 55 °C for 45 s, and 72 °C for 1 min, followed by 72 °C for 5 min. The PCR products were analyzed by standard agarose gel electrophoresis, and the bands were quantified with ImageQuant 5.0 software (Molecular Dynamics).
Statistical analysis
Data were expressed as the mean±SE. Comparison between groups was performed with the Student’s t test and χ2 analysis using a SigmaStat statistical software package (SPSS, Chicago, IL). P<0.05 was taken as showing significance.
Results
STAT3 mRNA and protein expression in Lewis lung cancer cells
STAT3 mRNA expression was determined using semiquantitative RT-PCR. After transfection of the cells for 72 h, there was no significant difference in STAT3 mRNA expression between vehicle and plasmid groups (Fig.l), showing that the plasmid vector itself had no effect on STAT3 mRNA expression. However, STAT3 mRNA expression in the STAT3 siRNA cells was significantly reduced (22%) compared to the two other groups (P<0.05, Fig.l).

Representative image of semi-quantitative RT-PCR analysis of Stat3 mRNA expression in the cells of the vehicle, plasmid, and STAT3 siRNA groups. RT-PCR was performed at 72 h after transfection. Lane M, marker; lane 1, vehicle; lane 2, STAT3 siRNA; lane 3, plasmid.
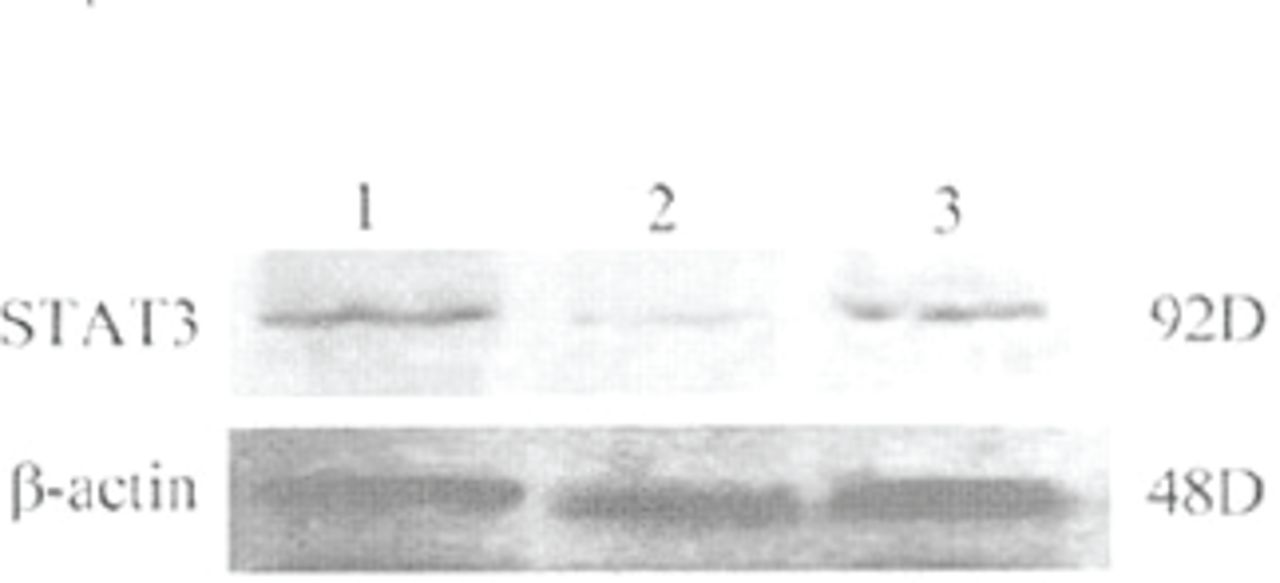
The level of STAT3 protein expression was evaluated using Western blot analysis with antibodies against the STAT3. Protein expression of the STAT3 protein in the vehicle group cells was not significantly different from the plasmid group cells at 72 h after transfection, showing that the plasmid vector itself had no effect on STAT3 protein expression. However, Fig.2 shows that treatment of the cells with STAT3 siRNA resulted in a significant decrease in the expression of the STAT3 protein compared to the two other groups (P<0.05).

Representative phothograph of Western blot analysis of Stat3 protein expression by in the cells of the vehicle, plasmid, and STAT3 siRNA groups. Western blot was performed at 72 h after transfection. Lane 1, vehicle; lane 2, STAT3 siRNA; lane 3, plasmid.
Proliferation of Lewis lung cancer cells
The MTT assay showed that the growth-inhibition rate of the STAT3 siRNA group (58%±3%) was significantly higher than that of the vehicle group (2.5% ± 0.8%; P<0.05) or plasmid group (2.1% ±0.5%; P < 0.05). However, growth-inhibition rates in the vehicle group cells did not significantly differ from the plasmid group, thereby indicating that the plasmid vector itself had no effect on proliferation of the cells.
Apoptosis of Lewis lung cancer cells
Flow cytometry and DNA laddering electrophoresis were adopted for determination of apoptosis. Flow cytometry analysis showed that the apoptotic-cell rate in the STAT3 siRNA group was significantly higher than that in the vehicle and plasmid groups (Table 1). However the apoptotic-cell rates in the vehicle and plasmid groups were not significantly different from each other (Table 1).
Flow cytometric analysis of apoptosis in the cells of the vehicle, plasmid, and STAT3 siRNA groups. Flow cytometry was performed at 72 h after transfection
The profile of DNA laddering electrophoresis is highly indicative of fragmentation of chromosomal DNA. As shown in Fig.3, by using this technique, transfection of the cells with STAT3 siRNA efficiently induced apoptosis. In contrast, cells in the vehicle and plasmid groups showed no significance of DNA frag mentation (Fig.3).
Representative image of an apoptotic ladder (indicative of fragmentation of chromosomal DNA) by agarose gel electrophoresis. Cells were from the vehicle, plasmid, and STAT3 siRNA groups. DNA laddering electrophoresis was performed at 72 h after transfection. M, marker; A, vehicle; B, plasmid; C, STAT3 siRNA.
Discussion
Lung cancer is the most frequent and one of the most deadly cancers with more than 1.1 million deaths annually worldwide.[35] The prognosis of this malignancy is still poor due to the absence of effective treatment methods. STAT3 has been recognized an important oncogenic transcription factor, thus it has the potential as a novel molecular target for treating lung cancers. [2, 21, 35] In our study with Lewis lung cancer cells, we found enhanced expression of the STAT3 gene at both mRNA and protein levels, promotion of cellular proliferation and inhibited apoptosis. On the other hand, inhibition of cellular STAT3 gene expression in vitro by STAT3 siRNA resulted in decreased expression of the STAT3 gene, inhibition of cell proliferation and induction of apoptosis.
Under physiological conditions (eg. in normal or benign tumor cells), STAT3 activation is transient and lasts from several minutes to several hours because of the transient nature of cytokine and growth factor signaling and the production of proteins that inhibit STAT3 expression. [5,36] However, numerous studies have indicated that aberrant STAT3 signaling might play an important role in the development and progression of a variety of cancers, [1,2, 12, 23, 30] Grandis et al.[12] have observed increased STAT3 activation in head and neck squamous cell carcinomas. Constitutive activation of STAT3 has been detected in many other cancers, such as multiple myeloma, skin, brain, breast, and lung cancers.[20, 23, 37, 38] To examine the expression of STAT3 gene in Lewis lung cancer cells, we used Western blots and RT-PCR analysis to measure STAT3 mRNA and protein expression showing that the cells displayed significant STAT3 gene expression. This finding is consistent with our previous studies and others studying many other cancer cells.[2, 3, 12] Additionally, in the present study, we determined whether STAT3 was associated with proliferation of the cells. Using the MTT assay to assay cellular proliferation, we demonstrated simultaneous enhanced cellular proliferation along with marked expression of the STAT3 gene, suggesting that increased expression of the STAT3 gene is related to proliferation of these cancer cells.
There is evidence demonstrating that constitutive activation of STAT3 can activate several genes whose products promote cell cycle progression and prevent apoptosis. [2, 12] Our previous studies and others have shown that increased STAT3 gene expression and decreased expression of Bcl-2 or Bcl-xL genes occur simultaneously in human laryngeal cancer cell lines (Hep2), prostate cancer cells, and in astrocytoma cells at both mRNA and protein levels.[2,3] In our study, Lewis lung cancer cells displayed inhibited apoptosis and others have suggested that malignant cells expressing continuously activated STAT3 become dependent on its activation for survival, and that disruption of activation or expression of STAT3 results in apoptosis. [2, 3, 30] Takgjj together, we hypothesize that STAT3 plays an important role in promoting Lewis lung cancer pro-liferation mediated by an anti-apoptotic pathway.
A variety of strategies for inhibiting STAT3 gene expression in cancers have been adopted. Leong et al. [30] reported that in human head and neck squamous carcinoma cells, blocking of STAT3 signaling by decoy oligonucleotides or antisense oligonucleotides nullified transforming growth factor effects and suppressed cell growth. It is noteworthy that siRNA is an alternative to the gene inhibition approaches mentioned above.[2, 3]
Theoretically, it is plausible that siRNA can be used to treat any disease linked to an overactive gene or genes with exquisite precision and high efficacy, by blocking the action of transcription factors and oncogenes with selective gene silencing.[27] Indeed, our previous studies and others have demonstrated that siRNA is effective in suppressing gene expression in vitro and/or in vivo in rats and mice. This therapy has achieved effective treatment of various organ and/or tissue disorders, including hepatitis, liver ischemiamperfusion injury, allograft rejection, central nervous system disorders and malignancies.[2, 3, 27, 39, 40]
There is evidence to suggest that siRNA targeting STAT3 (STAT3 siRNA) can serve as an effective anti-tumor approach to various malignancies.[2, 3,27] Konnikova et al.,[27] using STAT3 siRNA to silence STAT3 expression in human astrocytes and astrocytoma cell lines, reported that STAT3 regulates survivin expression and can induce apoptosis. Studies have demonstrated that blockade of STAT3 activation by STAT3 siRNA suppresses growth of human prostate cancer cells and induces apoptotic cell death.[41] In addition, our previous studies have demonstrated that inhibition of STAT3 gene expression by STAT3 siRNA results in inhibition of cell proliferation, and induction of apoptosis of human laryngeal cancer Hep2 cells in vitro and prostate carcinoma in vivo as evidenced by flow cytometric and TUNEL staining analysis of apoptosis.[2, 3] However, to date, these studies have not been conducted with human lung cancer tissues, so our study represents the first report using STAT3 siRNA with Lewis lung cancer cells. Transfection of the cells with STAT3 siRNA resulted in inhibition of STAT3 gene expression leading to a significant increase in the rate of growth inhibition, apoptosis, and DNA fragmentation compared to those cells treated by either culture medium or transfected with a control plasmid vector (Figs.1~3).
In conclusion, this study demonstrated that Lewis lung cancer cells display increased expression of the STAT3 gene accompanied by promoted cell proliferation and arrested apoptosis. Synthetic STAT3 siRNA can specifically inhibit STAT3 gene expression in these cells leading to growth suppression and induction of apoptosis. The use of siRNA therapy may provide a new treatment modality for lung cancer patients and other malignant tumors expressing constitutively activated STAT3.
Footnotes
This work was supported by the grant to from the Scientific and Technological Office of Jilin Province, China (No. 200505120).
- Received September 4, 2006.
- Accepted November 7, 2006.
- Copyright © 2006 by Tianjin Medical University Cancer Institute & Hospital and Springer
References
In this issue






{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.