

Abstract
Glioblastoma (GBM) is one of the deadliest tumors and has a median survival of 3 months if left untreated. Despite advances in rationally targeted pharmacological approaches, the clinical care of GBM remains palliative in intent. Since the majority of altered signaling cascades involved in cancer establishment and progression eventually affect cell cycle progression, an alternative approach for cancer therapy is to develop innovative compounds that block the activity of crucial molecules needed by tumor cells to complete cell division. In this context, we review promising ongoing and future strategies for GBM therapeutics aimed towards G2/M inhibition such as anti-microtubule agents and targeted therapy against G2/M regulators like cyclin-dependent kinases, Aurora inhibitors, PLK1, BUB, 1, and BUBR1, and survivin. Moreover, we also include investigational agents in the preclinical and early clinical settings. Although several drugs were shown to be gliotoxic, most of them have not yet entered therapeutic trials. The use of either single exposure or a combination with novel compounds may lead to treatment alternatives for GBM patients in the near future.
keywords
Introduction
Glioblastoma
For many years, tumors of the central nervous system (CNS) were primarily categorized according to histopathological criteria determined under microscopic observation, where similarities and phenotypic differences depended on cellular origin and their presumed levels of differentiation. In particular, gliomas are diffusely infiltrating glial cell tumors that are responsible for 80% of malignant tumors initiated in the brain and have been classified by the World Health Organization (WHO) depending on the type of the affected glial cell, thus integrating the nomenclature with grading system1. Histological diagnosis is correlated with tumor grade on a scale of I to IV based on how normal or abnormal the cells appear2. Accordingly, a grade IV astrocytoma (also called "glioblastoma", "glioblastoma multiforme", "grade IV glioblastoma", and "GBM") represents the most common and most aggressive primary malignant brain tumor, with 3 per 100,000 people diagnosed every year3. GBM is histopathologically characterized by brisk mitotic activity, cellular and nuclear atypia, vascular thrombosis, and microvascular hyperproliferation and necrosis, with 80% being primary or de novo occurring though malignant transformation from lower-grade gliomas (sometimes referred to as secondary GBMs)4.
GBM is one of the most deadly types of tumors5. If left untreated, this dismal tumor has a median survival of 3 months6. In addition to maximal safe surgical resection and radiotherapy (RT), the standard chemotherapeutic agent for its treatment since 2005 is the alkylant prodrug temozolomide (TMZ), which was first approved by the Food and Drug Administration (FDA) for use in recurrent GBM based on the phase II trial by Yung and colleagues7. Posteriorly, in the pivotal phase III study, Stupp and colleagues randomized ~600 patients diagnosed with GBM from various treatment centers. Their investigation consisted of radiation alone or radiotherapy with continuous daily TMZ, which demonstrated an improved 14.6-month median survival in the second group, versus 12.1 months in control patients. Two-year survival was also increased by 26.5% compared to 10.4% for those treated with radiotherapy alone. Nowadays, RT combined with concomitant and adjuvant TMZ after surgical resection, namely STUPP treatment, is widely used for newly diagnosed GBM patients8,9. Nonetheless, only 15%–20% of patients survive 5 years after diagnosis, and no other therapies have demonstrated a robust survival benefit in recurrent disease6,10.
TMZ is an imidazotetrazine derivative of the alkylating agent dacarbazine that delivers a methyl group to the purine bases of DNA (O6-guanine, N7-guanine, and N3-adenine). Although O6-methylguanine (O6-MeG) is the primary cytotoxic lesion, it can be reversed by the action of the repair enzyme methylguanine methyltransferase (MGMT), thereby neutralizing the cytotoxic effects of TMZ11. Accordingly, high expression of MGMT in glioma cells is the predominant mechanism underlying tumor resistance to alkylating agents12. Moreover, patients with methylated-MGMT treated with TMZ showed a 21.7-month median overall survival (OS) compared with 12.7 months in those with unmethylated promoters13, proving a direct association between MGMT expression and tumor response to TMZ therapy14.
Moreover, results from the European Organization for Research and Treatment of Cancer and National Cancer Institute of Canada trial recognized methylated-MGMT as the strongest predictor of outcome and benefit from TMZ treatment8,15. Similarly, the recent meta-analysis by Zhao and colleagues16 involving 7,886 patients, highlighted the universal predictive value of MGMT methylation in newly diagnosed GBM patients, elderly GBM patients, and recurrent GBM patients16.
Over the last two decades, many researchers have highlighted the importance of GBM molecular subtyping, but only recently was the WHO Classification for CNS Tumors able to integrate phenotypic and genotypic parameters, and subdivided GBM in three categories based on the status of the isocitrate dehydrogenase (IDH) gene17. Consequently, GBM are currently classified as IDH-wildtype (approximately 90% of cases that correspond most frequently to the clinically defined primary GBM), GBM IDH-mutant (approximately 10% of cases that closely correspond to the so-called secondary GBM), and GBM not otherwise specified (NOS), a diagnosis that is reserved for tumors without full IDH evaluation17. Importantly, some studies already have showed that OS of IDH-mutants are greater than IDH-wildtype gliomas18,19.
This current classification represents a conceptual and practical advance over its 2007 predecessor, reinforcing the need for molecular/genomic diagnosis, new molecular approaches, as well as further studies to gain a better understanding of the role of these mutational profiles in the survival of patients and their prognostic values.
Accordingly, many analyses of the genomic landscape of GBM were published by the Cancer Genome Atlas Research Network in 200820-22 and revealed specific genomic, epigenomic, transcriptomic, and proteomic alterations in core pathways that define novel specific tumor subgroups. Thus, some studies have predicted that genomic diagnoses will overrule and dictate the diagnosis in the future23. However, these subdivisions still play no role in current diagnostics and treatment decisions, but do help to overcome some of the molecular heterogeneity in GBM and improve treatment.
The concept of exploiting cell division as a therapeutic target has been in practice since the advent of chemotherapy. Briefly, antitumor treatments can affect the cell cycle through three modes of action: blocking DNA synthesis, causing DNA damage, or perturbing mitotic processes. As with many solid tumors, GBM is defined as a highly heterogeneous cancer with different cell populations coexisting within the tumor mass, each one with a distinct proliferative status directly connected to key molecules regulating cell cycle progression and mitosis. In this regard, the diagnostic and prognostic relevance of cell cycle biomarkers strongly reinforce the need to characterize signaling pathways and highlight their potential for novel targeted therapies for GBM. Hence, in the present review, we feature classic compounds used in anti-GBM therapy, and review recent advances on new therapeutic approaches that are based on the inhibition of cell cycle molecules.
Anti-microtubule agents
Mitosis disruption is widely used in clinics, and even with the rapid expansion in the number of classes of compounds with antineoplastic activity, anti-microtubule agents are still among the most strategic and have played a pivotal role in the curative and palliative treatment of cancer over the last 50 years.
In general, anti-microtubule compounds can be divided into tubulin destabilizing and stabilizing agents. In the first group, we can include vinblastine and vincristine, alkaloids isolated from the periwinkle plant Cantharanthus rosea, and colchicine, isolated from the meadow saffron Colchicum autumnale, all of which have been shown to inhibit microtubule assembly and to depolymerize steady state microtubules at substoichiometric concentrations24. In marked contrast to these classical agents, paclitaxel (the prototype of the taxane family), is another natural occurring anticancer agent originally isolated from the stem bark Taxus brevifolia that induces tubulin polymerization and forms extremely stable and nonfunctional microtubules, ultimately resulting in apoptosis25.
Microtubule-targeting drugs were long believed to induce cellular death by disrupting the spindle and delaying mitosis, however, it is now recognized that aside from their role in proper chromosome segregation, microtubules also play a significant role in many interphase functions, such as intracellular trafficking of proteins and organelles, migration, and maintenance of cellular shape. Consequently, microtubule interphase function impairment also brings an overall effect that ensures the efficacy of taxanes and vinca alkaloids26. Accordingly, agents targeting microtubule dynamics are widely used in the clinic, both alone and in combination with other chemotherapeutic agents, against multiple types of cancer. Nonetheless, these agents possess substantial liabilities and their use has been restricted by dose-limiting peripheral neurotoxicity27, severe myelosuppression28, and acquired resistance29-31.
In recent years, many new anti-microtubule drugs have emerged, most of which are still under clinical phase I/II trials26. Of note, a third-generation taxane cabazitaxel was approved in 2010 by FDA as the first therapy to show a survival benefit for the treatment of patients with docetaxel-refractory castration-resistant prostate cancer32. Moreover, the low solubility and susceptibility to elimination by multi drug resistance (MDR) pumps have triggered the development of alternative formulations. Nab-paclitaxel (Abraxane®), for instance, results from the covalent binding of albumin to the classical taxane to improve its cellular uptake and is currently being implemented in the treatment of pancreatic ductal adenocarcinoma33. Other innovative approaches that include the encapsulation of paclitaxel in nanocarrier systems, such as nanoparticles, liposomes, micelles, bioconjugates, or dendrimers34, hold promise to possess stronger antitumor activity and/or to minimize undesired side effects.
In line with this, new possibilities have emerged for GBM treatment (Table 1). As classical taxanes do not cross the blood-brain barrier (BBB)29, additional microtubule-stabilizing compounds such as epothilone D35 and dictyostatin36 are now showing therapeutic potential against GBM, with BBB permeability and slow brain clearance. Other strategies, including conjugates of paclitaxel and poly-L-glutamic acid37,38, paclitaxel delivered in nanoparticles39-43, or in biodegradable polyethylene-glycol filomicelles44,45, have also shown auspicious pre-clinical results.
Microtubule inhibitors and selective G2/M targeted compound tested in GBM in pre-clinical and therapeutic studies
Targeted therapy against G2/M regulators
The success of spindle poisons and the vulnerability of cancer cells to mitotic arrest have directed the search for new compounds to attain alternative mitotic targets (Figure 1), raising the necessity to overcome the limitations of tubulin-based antimitotic drugs and to expand the clinical effectiveness previously shown by these drugs. Currently, many of these selective drugs are being tested (Table 1).
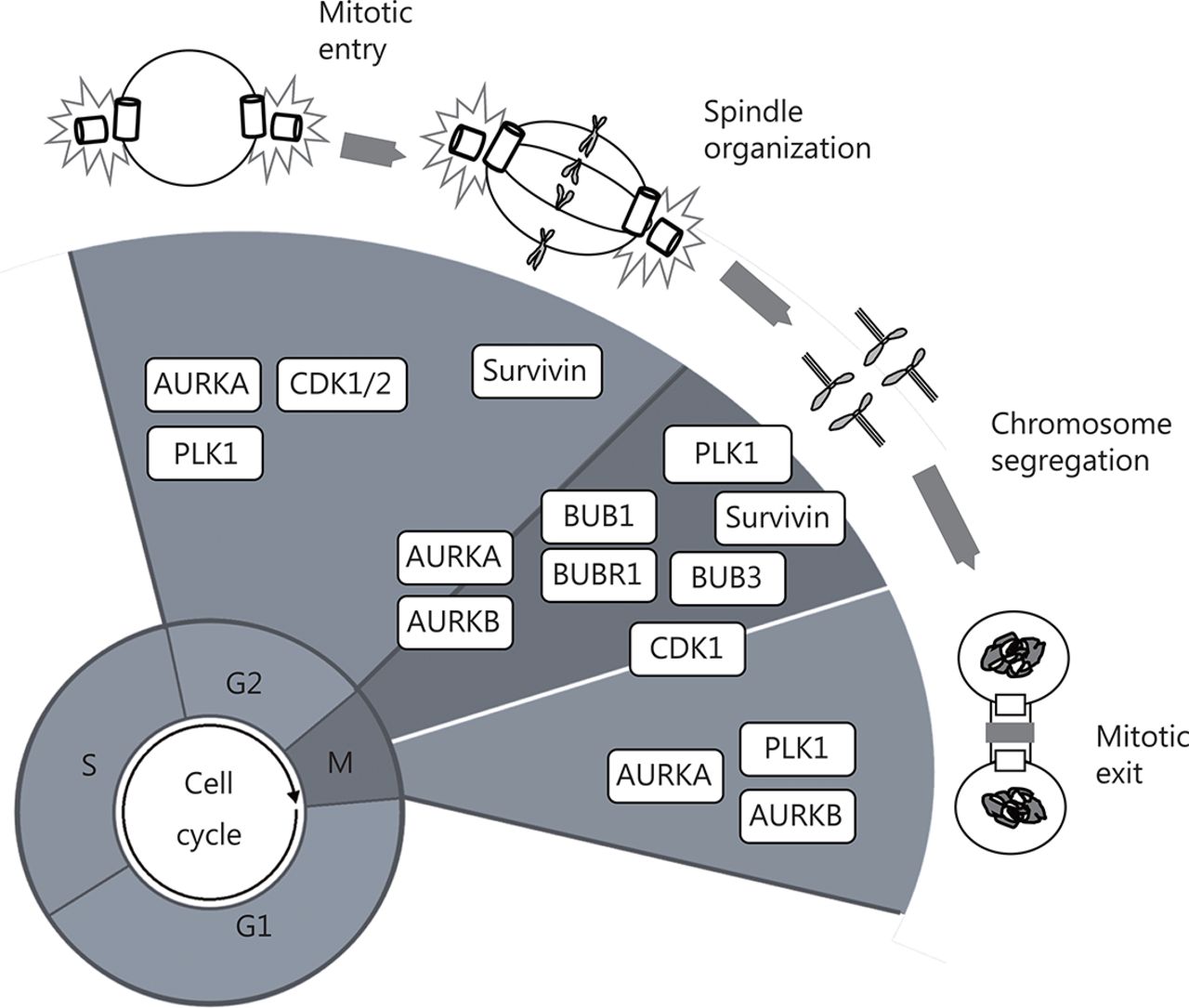
Schematic representation of plausible G2/M cell cycle regulators as therapeutic targets in GBM. CDK1 is essential for the G2–M transition and mitosis. BUBR1, BUB1, and BUB3 are fundamental to guarantee spindle assembly and correct chromosome separation by allowing their appropriate attachment to kinetochores. PLK1 is considered a master cell cycle regulator and is essential for the initiation of mitosis, centrosome maturation, bipolar spindle formation, cytokinesis, and the DNA damage response. AURKA and AURKB are crucial for spindle formation and guarantee the organization and alignment of the chromosomes during prometaphase, centrosome separation, and participate during cytokinesis. Survivin participates in cell division through its functions in the chromosomal passenger complex (CPC), spindle formation, checkpoint control, and assembling on polymerized microtubules.
Cyclin-dependent kinase (CDK) inhibitors
Cell cycle begins with a single cell that divides into two daughter cells with identical genomes107. This complex process is a well-ordered series of irreversible transitions from one state to the next. CDKs orchestrate progression through the consecutive phases, including entry into the cell cycle from quiescence, the G1/S phase transition, DNA replication in S phase, nuclear breakdown, chromosome condensation and segregation, and cytokinesis108.
Some CDKs (CDK1, CDK2, CDK3, CDK4, CDK6, CDK10, and CDK11) coordinate cell cycle regulation at different stages. CDK1 and CDK2, for instance, direct S and G2 phase transit, while CDK1 alone governs the G2/M transition and mitotic progression109. CDKs are generally activated when complexed with the adaptor molecules cyclins, but are also regulated by phosphorylation, interactions with inhibitory proteins, transient intracellular translocations, or by periodic proteolytic degradation of their activating cyclin partner. Other CDKs regulate cell cycle indirectly by activating other members of the family (CDK7, CDK20) or via transcription (CDK7, CDK8, CDK9, CDK19)110.
Amplification or mutation of cyclins, CDKs genes, or genes encoding their endogenous inhibitors, ultimately leads to deregulated CDK activity and loss of cell cycle control, a universal characteristic of cancer cells111-113. Changes in CDKs expression and/or regulation have been described in many human tumors, and occur frequently in the development of GBM114.
The search for CDK inhibitors began approximately 20 years ago and has led to the development of a plethora of compounds with variable selectivity that efficiently block cell-cycle progression and display potentially promising antitumor activities115,116. So far, the most thoroughly studied CDK inhibitors are flavopiridol and dinaciclib.
Flavopiridol is a semi-synthetic flavone that inhibits the activity of CDK1, CDK2, CDK4, and CDK6 by competitive binding at the adenosine triphosphate (ATP)-binding pocket117,118. In preclinical studies, its antitumor activity was rapidly recognized and confirmed in a variety of human tumor cell lines and xenograft models119,120. Specifically, in vitro testing of flavopiridol in GBM cell lines (including T98G, U87MG, U118MG, U251MG, and U373MG) induced apoptosis in a caspase-independent manner and independent of retinoblastoma and p53 tumor suppressor pathway alterations81. This drug also inhibited growth and cell migration in murine GBM, induced apoptosis, and reduced the expression of cyclin D1, CDK4, p21, and BCL-2. However, in contrast to reports on human cell lines, apoptosis was mediated by mitochondria121.
Nonetheless, despite its initial promise, flavopiridol has not displayed effectiveness in clinical trials as a single agent, and only marginal responses have been reported in the treatment of hematological malignancies122-124. This narrow therapeutic window and the identification of several off-target effects encouraged the development of the second-generation CDK inhibitor dinaciclib (SCH727665).
Dinaciclib inhibits CDK 1, 2, 5, and 9 with a better therapeutic index in preclinical studies125,126, and showed potent growth inhibiting activity in leukemia, osteosarcoma, and pancreatic cancer cells126-128. The drug also showed antitumor effects in a variety of human cell lines and xenograft models from the pediatric preclinical testing program129. Remarkably, treatment induced significant delays in event-free survival distribution compared to that of control treatment in more than 60% of evaluable solid tumors and in 3 of 7 leukemia xenografts, although poor results were obtained in the 3 xenograft GBM models129. Nevertheless, an initial clinical experience in patients with advanced leukemia showed transient cytoreductions and did not correlate with clinical outcome126. Many other compounds such as TG02130, P276-00131, SLM6132, and VMY-1-103133, among others, have been described to inhibit CDKs and may eventually enter clinical trials. Those previously tested in GBM are listed in Table 1.
Nonetheless, the inability of CDK inhibitors to produce prolonged remissions as single agents has increased the tendency towards their use in combination with other drugs. For instance, in vitro models have provided evidence of cooperative activity between flavopiridol and carboplatin in human ovarian cancer cells134, and between the CDK inhibitor and bortezomib in lymphoid and myeloid cell lines135,136. Moreover, flavopiridol has been shown to enhance TMZ-induced toxicity in human GBM cells and xenographic U87MG tumors in a p53-independent manner80, and to increase the pro-apoptotic and cytotoxic effects of ara-C in leukemia137. Early results in humans demonstrated 67% complete remission in patients with newly diagnosed acute myeloid leukemia (AML) after treatment with flavopiridol/cytarabine/mitoxantrone138. Auspicious results were also observed in a phase I trial of bortezomib and flavopiridol in patients with recurrent or refractory B-cell neoplasms139, providing additional prospects for pharmacological intervention.
Aurora kinases inhibitors
The accurate order of cell cycle progression events is ensured by tightly orchestrated feedback control mechanisms called "checkpoints", which prevent progression from phase to phase until particular critical conditions have been satisfied140,141. The metaphase checkpoint for example, also known as the spindle checkpoint, prevents the separation of chromosome or chromatid until they are properly attached to the spindle apparatus, guaranteeing correct chromosome placement on the metaphase plate and equitable chromosome segregation, resulting in the preservation of a stable diploid karyotype142. Conversely, abrogation of this mitotic checkpoint impairs the fidelity of chromosome segregation and induces chromosomal instability (CIN), a driving force of oncogenic transformation and tumor progression143,144.
The Aurora family of serine/threonine kinases includes Aurora A (AURKA), Aurora B (AURKB) and Aurora C (AURKC), all of which are essential for mitosis (AURKA and AURKB) and meiosis (AURKC) control145. Although the three AURK are involved in cell division, each one possesses specific functions. AURKA regulates the progression of mitosis by phosphorylating multiple substrates and promotes mitotic entry by governing the activation of Cyclin-B/CDK1145. Additionally, it controls centrosome maturation146, chromosome segregation, and bipolar spindle assembly147. AURKA expression, localization, and activity are consistent with its function as a centrosomal kinase. Specifically, there is an increase in its levels during the G2/M transition, and it is early localized at the centrosome and progressively associates with the mitotic poles and the adjacent spindle microtubules148.
On the other hand, AURKB is one of the most intensively studied kinases because it provides catalytic activity to the chromosome passenger complex (CPC). The CPC coordinates highly diversified processes, such as chromosome alignment, histone modification, and cytokinesis. For these reasons, AURKB is considered the "enzymatic heart" of this complex149,150. CPC can be considered as similar to the cyclin/CDK kinase complex, although instead of one nonenzymatic/regulatory subunit, the CPC contains three regulatory nonenzymatic subunits: survivin, borealin, and inner centromere protein (INCENP), all of which are subject to phosphorylation by AURKB151. AURKB kinase activity is critical for proper chromosome segregation150, initially through the phosphorylation of histone H3 on serine 10 (H3S10ph) to aid in mitotic chromosome condensation152,153. This kinase also contributes to the spindle checkpoint through phosphorylation of microtubule depolymerase mitotic centromere-associated kinesin (MCAK), which targets the protein to kinetochores, where it acts to mend any inadequate kinetochore attachment to the spindle154. Finally, AURKB is indispensable for accurate cytokinesis through phosphorylation of MgcRacGAP, a GTPase-activating protein, which is converted into RhoGAP and thus promotes cytokinesis155.
AURKC, on the other hand, appears to be the major enzymatic component of the CPC during meiosis, playing a specific role during female meiotic division and coordinating meiotic spindles in spermatogenesis156. Human AURKC is first expressed at the pericentric heterochromatin in pachytene spermatocytes157. In preimplantation embryos, it appears to be the major AURK expressed during the first three embryonic cell cycles, where it can be visualized on prometaphase chromosomes in zygotes and two- and four-cell-stage human embryos. Conversely, the endogenous AURKB protein is expressed at low-to-untraceable levels during these embryonic stages, but increases substantially after the eight-cell stage. It is interesting to highlight that the expression of AURKC occurs earlier, and is entirely substituted by AURKB at the blastocyst stage. Thus, it is tempting to hypothesize that AURKC could be the main enzymatic component of the CPC and thus, plays a specific role during human female meiosis and preimplantation embryo development157.
In terms of their role as mitotic regulators, deletion of AURKs could lead to cell division failure and embryonic development impairment. Increased expression or gene amplification of AURKs has been described in numerous cancers158, including breast159,160, ovarian161,162, gastric/gastrointestinal163,164, colorectal165,166, lung167,168, cervical169,170, prostate160,171,172, oral173,174, AML175,176, and glioma177,178. Importantly, in glioma tumors, AURKA and AURKB expression increases with tumor grade,179-182, and is significantly associated with GBM poorer patient survival183-185.
Many studies have confirmed the determinant role of AURKA in tumorigenesis through multiple mechanisms such as control of proliferation186, epithelial-mesenchymal transition (EMT)187, and metastasis188, as well as in the self-renewal capacity of cancer stem cells (CSCs)189. On the other hand, AURKB endorses cell cycle progression190 and the survival of cancer cells191, and AURKC kinase may encourage tumor progression160,192.
Thus, over the last decades, a series of AURK inhibitors have shown to effectively repress the progression and growth of many cancers (both in vivo and in vitro), suggesting that these kinases could represent novel therapeutic targets.
ZM447439, a selective ATP-competitor, was reported as the first Aurora kinase inhibitor in 2003193. This compound inhibits the phosphorylation of histone H3 on serine 10, a physiological target of AURKB194, causing AURKB to be more selectively inhibited both in vitro and in vivo compared to AURKA195. Borges et al.106 found that this inhibitor decreased proliferation and acted synergistically with TMZ in primary cultures and cell lines of GBM, while inducing apoptotic cell death.
Moreover, MLN8054, a small-molecule inhibitor of AURKA, induces accumulation of cells in G2/M phase and spindle defects in vitro196, leading to aneuploidy through chromosome congressional defects197. This compound was administered orally to breast, colon, pancreatic, and bladder cancer patients198 in a phase I clinical trial via capsules (5 or 25 mg) once daily for 7 uninterrupted days every 21 days and was later extended to evaluate increasing durations of oral dosing in patients with advanced malignancies199. The main side effect was grade 2/3 somnolence, which was attributed to the binding of the agent to the gamma-aminobutyric acid α-1 benzodiazepine (GABAAα1 BZD) receptor. Posteriorly, MLN8237 (alisertib), a more potent inhibitor was introduced and the structural modification of a methoxy group to either end of the MLN8054 molecule resulted in less benzodiazepine-like side effects200. This second generation inhibitor readily crosses the BBB and acts as a specific AURKA inhibitor at concentrations lower than and equal to the maximally tolerated dose in animal models and is currently being tested in a variety of phase I-II clinical trials201. Of note, two independent groups showed that MLN8237 exhibited potent effectiveness against glioblastoma neurosphere tumor stem-like cells in vitro and protracted the median survival of mice bearing intracranial human GBM neurosphere tumor xenografts202, while potentiating the effects of TMZ100.
Another interesting compound VX 680, also known as MK-0457, tozasertib, or VE465, is a synthetic, pyrimidine derivative with affinity for AURKA, AURKAB, and AURKAC. Nanomolar concentrations of MK-0457 show powerful antitumor activity by inhibiting phosphorylation of histone H3, causing accumulation of cells with 4N DNA, thus preventing cytokinesis and inducing massive apoptosis in various cancer cell lines. In preclinical models, MK-0457 has been shown to impede tumor xenograft growth and prompt tumor regression203. Moreover, in its first phase I clinical trial, intravenous continuous infusion of MK-0457 was given over several days to patients that had been previously treated for solid tumors204, and was found to show activity in a phase II study, administrated to patients with Philadelphia chromosome-positive acute leukemia205.
In GBM cells, MK-0457 decreased colony formation, increased polyploidy, and p53 expression, resulting in cell growth inhibition in a caspase-independent manner105. In parallel, AZD1152, a quinazoline prodrug, showed high affinity for AURKB and AURKC, with comparable effects to MK-0457 in cancer cells206. Specifically in GBM, AZD1152 induced polyploidy and non-apoptotic cell death regardless of p53 status and was accompanied by poly-merotelic kinetochore-microtubule attachments and DNA damage in all the cell lines tested102. Moreover, AZD1152 treatment enhanced the expression levels of the death receptor TRAIL-R2, thus increasing the natural killer (NK) cell ligand MIC A/B in p53-deficient cells, along with an induction of FAS/CD95 in p53-proficient cells leading to NK-cell-mediated lysis, thus highlight a p53-independent mode of action102. Additional AURK inhibitors that are less well studied but have been tested in GBM are listed in Table 1.
PLK1 inhibitors
Polo-like kinase (PLK) 1 is the most well studied member of the PLK family. This serine threonine kinase has a highly conserved N-terminal Ser/Thr kinase catalytic domain and plays a critical role in multiple steps of cell cycle progression and the DNA damage response207. Recently, PLK1 has emerged as a potential target for cancer therapy, as its overexpression is prevalent in various malignant tumor types and acts as a prognostic factor208,209.
This master cell cycle checkpoint protein shows its expression peak at G2/M phase and is involved in mitosis initiation, centrosome maturation, bipolar spindle formation, cytokinesis, and participates during the DNA damage response207. Preclinical studies in vitro and in vivo have shown that its blockage has a significant impact and can be explored for cancer treatment of both solid and hematologic malignancies.
Several anticancer drug candidates targeting PLK1 have been developed, and some agents have shown auspicious outcomes in early-phase clinical trials, though none of them have achieved clinical applications. PLK1 inhibitors can be classified according to their mode of action, even though most of them competitively bind to the ATP-binding site such as BI 2536, BI 6727, and GSK461364210. However, there are other inhibitors that target regions outside the ATP pocket, such as ONO1910, and those that can even bind to PLK1 through the PDB domain210,211.
In general, PLK1 inhibitors have shown favorable pharmacokinetic profiles, safety, and efficacy in patients with solid tumors. BI 2536, the first PLK1 inhibitor to be tested as monotherapy in humans, is currently being explored in a combinational study conducted in patients with diverse solid tumors212. Nonetheless, most data from clinical studies reported several adverse effects on patients including neutropaenia, thrombocytopaenia, anaemia, and pain213. Contrarily, the second-generation BI 6727 (also known as volasertib) showed improved pharmacokinetic profile, safety, and efficacy in phase II studies. Of note, the benefits obtained by treating AML with volasertib awarded the Breakthrough Therapy status for this drug by the FDA214, though its adverse effects are still an important issue to be considered . Comparatively, a recent phase I dose escalation study with NMS-1286937 successfully identified the maximum tolerated dose and toxicity, but even with disease stabilization in several patients, hematological toxicity was also limiting215.
Other multi-tyrosine kinase inhibitors, such as pazopanib and dasatinib, that are known to inhibit PLK1 at high concentrations, have been approved for therapeutic use by the FDA216,217 despite the fact that they may not inhibit PLK1 activity in a direct manner217.
On the other hand, PLK1 inhibitors have shown to be more effective when other genetic alterations are present in cancer cells. For instance, the thiophene benzimidazole GSK461364 has shown a superior antitumor effect in p53-mutated tumors in a preclinical study, although it must be co-administered with anticoagulants because of the high occurrence of venous thrombotic emboli212. Others, like RO3280, poloxin, and ON 01910, that are also being tested in vitro218, have shown to cause mitotic impairment and apoptosis in cancer cells219,220.
In GBM, BI 2536 has shown to cause G2 arrest, restrain cell proliferation and survival, and even synergize when combined with TMZ treatment71,221. Furthermore, when analyzing the CD133(+) tumor-propagating cells from primary GBM, a high level of PLK1 has been shown and its inhibition has an antitumor effect when combined with anti-BRAF drugs222. Other compounds like J10198409 have also shown to disrupt GBM stem cell proliferation70, and are sensitive to TMZ (Table 1).
BUB1 and BUBR1 inhibition
The mitotic spindle checkpoint (also known as spindle assembly checkpoint or SAC) is one of most important cell controls to guarantee the correct segregation of chromosomes141. Any dysregulation of SAC genes expression leads to DNA aneuploidy223. Several genes have been identified as part of this process, among which the budding uninhibited by benzimidazole (BUB) and the mitotic arrest deficient (MAD, also known in humans as BUBR1) family members are the most explored224,225.
The BUB family includes BUB1 and BUB3, while the BUBR1 family is composed by MAD1, MAD2, and MAD3. These kinases ensure correct chromosome segregation by playing key roles in averting the premature separation of sister chromatids until all chromosomes are appropriately attached to kinetochores226. BUBR1 is not only required for spindle checkpoint, but is also needed for chromosome alignment227. BUB1/BUBR1 and MAD2 operate as elements of distinct pathways sensing tension and attachment.
Variation on SAC gene expression was reported in human CNS tumors228. Alterations of different mitotic checkpoint proteins are important for GBM development and maintenance, and their levels are frequently inversely correlated to prognosis. Recently, it was shown that senescent GBM cells have aberrant centrosome morphology, and depletion of protein kinase C, which is fundamental to induce mitotic slippage-induced senescence229. Moreover, other kinases such as the monopolar spindle 1 (MSP1) are also important for spindle integrity and have shown to be regulated by the miR-21 in GMB cells230.
Mutations on BUB1, BUB3, and BUBR1 do not play substantial roles in the causation of chromosomal instability in GBM231. Nonetheless, a correlation between glioma grade and expression level of SAC genes was already reported, and BUB1 was associated with survival rates and proposed as a survival predictor232. Upregulation of BUB1 and BUBR1 expression and the downregulation of BUB3 were described in GBM samples and cell lines. Moreover, inhibition of BUB1 and BUBR1 via siRNA has shown to be efficient in decreasing cell proliferation and colony formation and, when combined with other drugs such as TMZ, increasing cell cycle arrest and apoptosis233.
In this way, SAC proteins are considered viable options for GBM therapy, though only few inhibitors are currently available, and none of them are FDA approved. Inhibitors of MSP-1 have already been tested in vitro showing promising results by sensitizing GBM cells to anticancer drugs234. The BUB1 inhibitors cycloalkenepyrazoles235, BAY-320 and BAY-524 have also shown to be efficient to sensitize cells to treatment with paclitaxel by compromising chromosome segregation and cell proliferation236.
Survivin
Survivin (also known as BIRC5) is a member of the inhibitor of apoptosis (IAP) family with key roles in the control of cell division and inhibition of apoptosis237-239. Of all the IAPs, survivin is the smallest with a single N-terminus BIR domain and C-terminus coiled coil (CC) domain and six alternative splicing variants or isoforms described until today240: wildtype (WT), 2B, ΔEx3, 3B, 2α, and 3α. Of those, survivin WT, 2B, and ΔEx3 variants have been extensively investigated for clinical and prognostic associations in cancer241. Notably, the role of novel isoforms in the regulation of apoptosis shows conservation of anti-apoptotic properties for survivin-WT and –ΔEx3 variants and a markedly reduced anti-apoptotic potential for survivin-2B242.
Functionally, survivin is considered a nodal protein, meaning that it is a protein involved in multiple signaling mechanisms in tumor maintenance and interacts with a large number of molecules, regulators, transcriptional networks, and modifiers that are involved in its functions, either directly or indirectly243. Consequently, it is possible to include survivin in multiple cellular networks. It participates in cell division through its functions in the chromosomal passenger complex (CPC), spindle formation, checkpoint control, and assembling on polymerized microtubules. Furthermore, in the anti-apoptotic network, survivin provides a heightened cell survival threshold and cooperates intermolecularly with adaptor or cofactor molecules of the cytosol and mitochondria243. Interestingly, increasing evidence indicates that survivin restricts autophagy, and its downregulation may induce apoptosis through autophagy-dependent mechanisms, including interactions with the autophagy regulator beclin 1244. In addition, recent reports show that in cancer cells, the translocation of survivin into the nucleus may increase DNA repair by upregulating Ku70245. Furthermore, survivin forms a complex with Ku70 and γH2AX in cells following irradiation246, and it has been widely demonstrated that its overexpression causes resistance to various chemotherapeutic (vincristine, cisplatin, bortezomib, tamoxifen) and pro-apoptotic agents (TNF-α, TRAIL)247-253. Of note, survivin is almost untraceable in normal adult tissues, but presents very high and unique expression patterns in most human tumors, making it an ideal selective target for cancer treatment243. Moreover, its plethora of functions allows for the disruption of numerous tumor-promoting networks with global anti-proliferative effects. For these reasons, many compounds have been developed to block its transcription, translation, and function in tumor cells243.
In gliomas, particularly GBM254, the high levels of survivin have been strongly related with a poor prognosis and resistance to chemotherapy and radiotherapy255,256. Among the more well studied survivin antagonists, the small molecules 1-(2-methoxyethyl)-2-methyl-4,9-dioxo-3-(pyrazin-2-ylmethyl)-4,9-dihydro-1H-naphtho (2,3-d) imidazolium bromide (YM155) and tetra-O-methyl nordihydroguaiaretic acid (M4N) have already been tested in GBM cells. YM155, specifically inhibits survivin gene promoter activity257 and its anti-neoplastic effect have now been reported in GBM cell lines with normal or deficient DNA-dependent protein kinase activity258. In addition, this drug caused a drastic decrease in the invasive and metastatic capacities of the GBM cells259, However, phase I and II trials have shown modest efficacy260,261.
On the other hand, M4N can inhibit the transcription of survivin by interfering with the binding of the Sp1 transcription factor to its promoter262. Recently, Castro-Gamero et al.263 showed that M4N treatment downregulated the expression of survivin and the survivin-ΔEx3 variant, and augmented the levels of survivin-2B variant, while decreasing cell proliferation, inducing apoptosis and acting synergistically with chemotherapy in GBM primary cultures and cell lines. Comparable to YM155, M4N has also been included in clinical trials. Safe profiles and partial responses have been demonstrated in a few patients with chronic myeloid leukemia (CML) or AML264. Interestingly, in a phase I study that included high-grade glioma patients, the safe daily dose was established as 1,700 mg, although the long-term stability and the lack of associated myelosuppression suggested that M4N could be safely combined with radiation and TMZ in newly diagnosed high-grade gliomas265.
On the other hand, while targeting the transcription of survivin is already being tested in clinical trials, YM155 and M4N are not selective and may also act on one or more upstream transcription factors that regulate the expression of many other downstream genes. Consequently, designating these compounds as specific survivin inhibitors is difficult266.
G2/M inhibition and GBM standard treatment
As previously described, TMZ is the first line drug for GBM treatment. The drug was initially approved in the 1980s for grade III gliomas. Due to its oral administration and ability to cross the BBB, TMZ was rapidly incorporated into GBM treatment. Nonetheless, its efficacy is hampered by the presence of active MGMT and has only brought mild improvements to treatment efficacy, even in patients that do not express the DNA repair protein. Thus, there is a constant search for drugs to potentiate its cytotoxic effects. Within this review, we have highlighted G2/M inhibitors that have already shown synergistic effects with TMZ in GBM pre-clinical studies and summarized these findings in Table 2.
G2/M inhibitors that have proven synergistic effects with GBM standard treatments (TMZ and radioitherapy) and other drugs in vitro
Radiotherapy, on the other hand, is considered one of most effective cancer treatments after surgery. However, despite the combination of treatment strategies, GBM prognosis remains low. Several drug combinations have been proposed to increase radiotherapy response. Many of the cell cycle inhibitors cited above have proven encouraging results in vitro (Table 2).
PLK1 blockage by BI 2536, has a radiosensitizing effect on GBM by causing a G2/M arrest and leading to an increase in cell death71. Similar results were seen when another PLK1 inhibitor (GSK461364) was used75.
Similarly, Lehman et al.182 found that MLN8237 was potently cytotoxic and sensitized GBM cells to ionizing radiation in vitro through AURKA inhibition. Moreover, this drug inhibited the proliferation of GBM neurospheres and potentiated the effects of TMZ and ionizing radiation via inhibition of phosphor-Thr(288) AURKA100. ZM447439 (associated with TMZ) also enhanced the effects of radiation in GBM cells106. Furthermore, YM155 has shown to increase the percentage of giant multinucleated cells and centrosomal overduplication of U87 cells after irradiation, causing mitotic cell death267. Similar results were obtained after treatment of GBM cells with M4N263.
Collectively, these findings highlight the potential of using cell cycle proteins for improved outcome of GBM by enhancing the response to radiation treatment.
Conclusions
Targeting the cell cycle machinery, especially mitotic proteins, continues to hold great promise as a potential strategy to combat cancer progression, showing very encouraging results in the preclinical setting. At present, a plethora of drugs have proven to be gliotoxic, but only a small number have entered therapeutic trials and from those, even fewer have proven to have effective responses (Figure 2). Nonetheless, many compounds are being tested for GBM and in the near future, single exposure or combinations may lead to treatment alternatives for patients with this devastating tumor.

G2/M inhibitors that have been tested in patients with GBM. Formulas were obtained at the NIH Pubchem Open Chemistry Database (https://pubchem.ncbi.nlm.nih.gov).
Acknowledgements
This work was supported by The São Paulo Research Foundation – FAPESP (Grant No. 12/16888-8 and 2009/50118-2).
Footnotes
Conflict of interest statement No potential conflicts of interest are disclosed.
- Received February 19, 2018.
- Accepted June 26, 2018.
- Copyright: © 2018, Cancer Biology & Medicine
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY) 4.0, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.
- 41.
- 42.
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.
- 48.
- 49.
- 50.
- 51.
- 52.
- 53.
- 54.
- 55.
- 56.
- 57.
- 58.
- 59.
- 60.
- 61.
- 62.
- 63.
- 64.
- 65.
- 66.
- 67.
- 68.
- 69.
- 70.↵
- 71.↵
- 72.
- 73.
- 74.
- 75.↵
- 76.
- 77.
- 78.
- 79.
- 80.↵
- 81.↵
- 82.
- 83.
- 84.
- 85.
- 86.
- 87.
- 88.
- 89.
- 90.
- 91.
- 92.
- 93.
- 94.
- 95.
- 96.
- 97.
- 98.
- 99.
- 100.↵
- 101.
- 102.↵
- 103.
- 104.
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.
- 124.↵
- 125.↵
- 126.↵
- 127.
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.↵
- 174.↵
- 175.↵
- 176.↵
- 177.↵
- 178.↵
- 179.↵
- 180.
- 181.
- 182.↵
- 183.↵
- 184.
- 185.↵
- 186.↵
- 187.↵
- 188.↵
- 189.↵
- 190.↵
- 191.↵
- 192.↵
- 193.↵
- 194.↵
- 195.↵
- 196.↵
- 197.↵
- 198.↵
- 199.↵
- 200.↵
- 201.↵
- 202.↵
- 203.↵
- 204.↵
- 205.↵
- 206.↵
- 207.↵
- 208.↵
- 209.↵
- 210.↵
- 211.↵
- 212.↵
- 213.↵
- 214.↵
- 215.↵
- 216.↵
- 217.↵
- 218.↵
- 219.↵
- 220.↵
- 221.↵
- 222.↵
- 223.↵
- 224.↵
- 225.↵
- 226.↵
- 227.↵
- 228.↵
- 229.↵
- 230.↵
- 231.↵
- 232.↵
- 233.↵
- 234.↵
- 235.↵
- 236.↵
- 237.↵
- 238.
- 239.↵
- 240.↵
- 241.↵
- 242.↵
- 243.↵
- 244.↵
- 245.↵
- 246.↵
- 247.↵
- 248.
- 249.
- 250.
- 251.
- 252.
- 253.↵
- 254.↵
- 255.↵
- 256.↵
- 257.↵
- 258.↵
- 259.↵
- 260.↵
- 261.↵
- 262.↵
- 263.↵
- 264.↵
- 265.↵
- 266.↵
- 267.↵
In this issue






{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- HeberFERON distinctively targets Cell Cycle in the glioblastoma-derived cell line U-87MG
- Anti-tumor pharmacology of natural products targeting mitosis
- Thiabendazole Inhibits Glioblastoma Cell Proliferation and Invasion Targeting Mini-chromosome Maintenance Protein 2
- Omics-based integrated analysis identified ATRX as a biomarker associated with glioma diagnosis and prognosis